Critical Rashes to Identify in the Emergency Department
Critical Rashes to Identify in the Emergency Department
Authors: Joshua L. Wright, MD, Military Component Program Director for Emergency Medicine, Assistant Professor, of Emergency Medicine, Boonshoft School of Medicine, Wright State University, Dayton, Ohio; Thomas Therrien, DO, Resident Physician, Emergency Medicine Residency, Boonshoft School of Medicine, Wright State University, Dayton, Ohio; Jonathan I. Singer, MD, FAAP, FACEP, Associate Program Director for Emergency Medicine, Professor of Emergency Medicine and Pediatrics, Boonshoft School of Medicine, Wright State University, Dayton, Ohio.
Peer reviewer: George L. Foltin, MD, FAAP, FACEP, Associate Professor of Pediatric and Emergency Medicine, New York University School of Medicine, New York, New York.
Rashes are common in the emergency department and may be a challenge diagnostically. The authors review rashes that the clinician cannot afford to miss.
— The Editor
Introduction
Skin disorders are encountered frequently in the emergency setting. In general (in non-pediatric emergency departments [EDs]), it is estimated that 4% of pediatric chief complaints are dermatologic and1 in dedicated pediatric ED's they account for 5-31% of visits.2,3 Fortunately, a majority of skin disorders are not medical emergencies. However, there are a small number of illnesses with prominent skin manifestations that are important causes of morbidity and mortality. These conditions require vigilance to make an early accurate diagnosis and minimize morbidity and mortality.
Emergency physicians have long been advised to approach the differential diagnosis of dermatologic conditions based on the appearance of skin lesions.4 This descriptive approach is also effective for identification of critical rashes. One model notes six, broad morphologic categories of dermatologic life threats. The categories are: petechial/purpuric, maculopapular, ulcerative/pustular, vesicular/blistering, serpiginous, and mixed. A critical rash for each category is reviewed in this article.
Petechia/Purpura
Meningococcal Infection. The bacteria, Neisseria meningitidis, silently colonizes the nasopharynx of 5-15% of the general population and 2-3% of the pediatric population and is transferred in droplets by close exposure. Carriage generally leads to the formation of protective antibodies. Maximal progressive increase in the development of antibodies occurs between the ages of 2-12 years. Only an exceedingly small number of susceptible or recently-colonized individuals will develop any symptoms. It is postulated that a viral upper respiratory infection or passive exposure to tobacco fumes damages the nasopharyngeal epithelium and facilitates bacterial translocation to the local tissues. Bloodstream invasion may follow. In non-epidemic situations, previously-healthy children younger than age 2 are most susceptible to bloodborne meningococcal infection. In epidemics, school-aged children, including adolescents, are more often affected. Individuals of all ages with complement component or alternative pathway (properdin) deficiencies are at increased risk for fulminant disease.5
Infection from Neisseria meningitidis causes a wide spectrum of disease. The illness ranges from acute to subacute and non-life threatening to life and limb threatening. In the acute, non-life threatening state, the organism causes a nonspecific, brief febrile illness that can be associated with positive blood cultures.6 Despite the occult bacteremia, the illness is transient. Other self-limited, non-life threatening infections are localized. Specific body parts that are affected include the conjunctivae; ocular globe; pharynx; bronchus; larynx; joints; middle ear; lung; and genitourinary tract, including the prostate, cervix, and urethra. Potential life threatening, localized sites of infection include the pericardium and meninges. In the non-life threatening, subacute expression of the illness, Neisseria meningitidis can be recovered over days and weeks from the bloodstream in patients who have fever and arthralgia (chronic meningococcemia). Although often untreated, these patients usually do well. This is in contrast to the patient who goes from an asymptomatic state to near death within a hyperacute timeframe from bloodstream invasion (acute meningococcemia).
Acute meningococcemia causes a relatively uniform, classic presentation (see Table 1).7,8 Acute meningococcemia is associated with a sudden fever spike, generally in the 39-40.5°C range. Temperature elevation is the first symptom in one-half of the cases.9 Muscle aches are common and myalgia may be severe enough that children refuse to walk.10 Parents of preverbal children note poor feeding, irritability, and alteration in mental status ranging from mild somnolence to frank lethargy.
| Table 1. Clinical Symptoms and Their Frequencies in Meningococcemia |
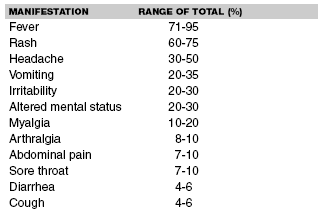 |
Generalized petechiae and purpura are prominent features of fulminant meningococcal disease, with little or no meningeal involvement. Petechiae alone are present in approximately 50% of individuals with meningococcemia.11 The number of petechiae range from fewer than a dozen to too-numerous-to-count.12 The petechiae are typically found on the extremities and trunk with greater frequency, and can, in isolated circumstances, appear on the palms, soles, anterior thorax, or perioral and mucous membranes. The petechiae also may be associated with a palpable, purpuric eruption. Purpura alone is noted in approximately one-quarter of patients.13 Pathologically, purpura results from microvascular thrombosis, capillary leak, and hemorrhage into the skin and soft tissue structures. (See Figure 1.) Large ecchymotic areas, particularly in the distal extremities, including the digits and distal facial structures such as earlobes and nasal tip, may become gangrenous.
| Figure 1. Petechiae and Purpura Associated with Meningococcemia |
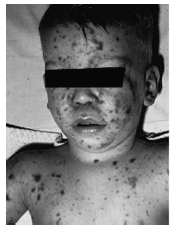 |
Four uncommon dermatologic manifestations of meningococcemia may occur. The least common is a large erythematous tender patch that blanches, resembling erysipelas.14 Another uncommon variation appears to be a diffusely urticarial rash. A third variant is a pink, raised, almost vesicular eruption that resembles early varicella. The final atypical rash is macular or maculopapular lesions on the trunk and extremities that resemble many common viral exanthems.12
Bedside tests can be performed by the emergency physician and may support the provisional diagnosis of meningococcal sepsis. However, fewer EDs are now performing bedside tests that are not CLIA waived, so this may not be an available option. The first test requires filling a hematocrit tube, spinning the specimen, and Wright staining or gram staining the buffy coat. A leukocyte-rich smear of peripheral blood is a rapid test for diagnosing Neisseria meningitidis bacteremia. Positive rates of recovery are different depending upon the degree of qualitative bacteremia. The second microbiologic test requires applying the serosanguinous fluid that is released as a petechial or purpuric lesion is abraded to a glass slide. Following air drying and heat fixing, a gram, Wright, or methylene blue stain may reveal the offending organism when examined with oil immersion. As an alternate, a gram stain aspirate of a skin lesion is positive in 50-70% of proven meningococcal infections.15 Latex agglutination kits for the detection of bacterial polysaccharide antigens to Neisseria meningitidis from blood and urine provide too high a rate of false-positives and false-negatives to be of utility.16 There are currently no reliable commercial polymerase chain reaction (PCR) tests of blood or urine for the detection of N. meningitidis.
The differential diagnosis for febrile childhood petechiae with or without purpura includes a modest number of diseases. The conditions are traditionally separated pathophysiologically into acquired vascular, coagulation cascade, or platelet disorders.17 An effective ED strategy is to separate the conditions by the presence or absence of toxic appearance. (See Table 2.) Several infectious agents may cause ill appearance, fever accompanied by rapidly-progressive hemorrhagic necrosis of the skin, and involvement of visceral organs such as kidneys and adrenal glands. Similar multisystem manifestations are seen with acute bacterial sepsis from several species. Streptococcus pneumoniae and Haemophilus influenzae are less common in the post Hib (Haemophilus influenzae type b) and Prevnar vaccination era.18,19 Several viral infections produce an acute febrile illness with prostration, headache, myalgia, nausea, vomiting, and a hemorrhagic diathesis. Some of the arboviruses (dengue), arenaviruses (lassa), and Bunyaviridae family (hantavirus) are notorious for causing acute multisystem disease followed by petechiae and purpura. These acute viral infectious agents carry a high mortality rate and a high morbidity rate secondary to skin and tissue loss in survivors. Before the physician considers these agents, a history of travel by the patient must be confirmed.20 Confirming travel to an endemic area also is necessary before considering rickettsial illnesses or malaria. Purpura fulminans refers to the circumstance of deterioration 7-10 days after an acute viral or bacterial infection. The affected patient typically has an acute illness such as varicella or group A beta-hemolytic streptococcal pharyngitis. During their recovery, there are systemic manifestations followed by circumscribed ecchymosis of the skin. The purpuric lesions primarily involve the trunk. There is usually sparing of extremities and lesser involvement of the kidneys, adrenal glands, and lungs.21
The principle goals of ED management of apparent sepsis with coagulopathy are to confirm adequate airway, breathing, and circulation; define the underlying cause; and limit morbidity by reversing expected imbalances. Aggressive fluid delivery, pressor agents, blood derivatives, and broad-spectrum antibiotics are non-controversial therapeutics for apparent meningococcemia. The role for low-dose, unfractionated heparin,22 hyperbaric oxygen,23 and corticosteroids24,25 remains controversial.
Maculopapular
Rocky Mountain Spotted Fever. Rocky Mountain spotted fever (RMSF) is a tick-borne infection caused by the bacterium Rickettsia rickettsiae. It is one of the most common vector-borne illnesses in the United States. The incidence is highest among children. RMSF is associated with mortality, causing approximately 37 deaths in the United States each year.26 Fatality rates are highest among males and in adults.27 While there have been reported cases in every month of the year, most cases occur from April to September. RMSF is endemic from Canada through Central America, with some cases being reported in Brazil and Columbia.28
RMSF has a wide clinical range that spans from a mild, undifferentiated febrile illness to fulminant disease leading to death. One should be suspicious of the disease when illness follows a tick bite. Unfortunately, 30-50% of patients do not have a history of tick bite. Most diagnosed patients report residing in or traveling to an area with known tick infestation. The first symptoms occur from 2 to 7 days after the patient is bitten by an infected tick. An insidious malaise and diffuse lumbar, thigh, and calf myalgia herald the onset of the disease. Fever follows with either a gradual rise or abrupt spike. Fever persists for several days and may oscillate with antipyretic therapy. Usually, temperatures remain persistently above 39°C and often ascend to the 40-40.6°C range. Bifrontal and retroorbital headache are exacerbated during heightened temperatures, but severe headaches persist when fever is lowered.29 Gastrointestinal symptoms occur in one-third of patients and include anorexia, nausea, vomiting, diarrhea, and generalized abdominal pain.30 Within a span of 2-6 days from the onset of symptoms, skin eruption occurs.
A rash occurs in RMSF in the majority of cases. The rash, when present, is "classic" in 50-80% of cases both in morphology and distribution. At onset the rash resembles delicate, pink macules which easily blanch with pressure. Within a day, the macules become maculopapular and are less likely to blanch. After several days, the longest existing lesions transition to non-blanching petechiae. The lesions first appear on the periphery, especially on the wrists and ankles. The rash spreads centripetally up the extremities to the trunk (see Figure 2), and then becomes confluent. Facial, interdigital, webspace, palm, and sole involvement occurs last. In atypical circumstances, the acral or truncal rash becomes confluently hemorrhagic, progressing to skin necrosis.31 In 10-15% of laboratory confirmed cases of RMSF, the rash is never seen.
| Figure 2. Centripetally Spreading Maculopapular Truncal Eruption of RMSF |
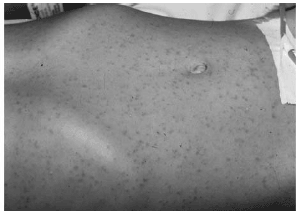 |
No laboratory tests with rapid turnaround time are available to the emergency physician to confirm RMSF. Characteristic hematologic and serum changes of RMSF fever can add credence to the provisional diagnosis. Complete blood counts between the fourth and seventh day of the illness show mild leukopenia or normal total white blood cell count with neutrophilic predominance and thrombocytopenia of varying severity.32 For unexplained reasons, hyponatremia is seen in many patients with RMSF. When combined with minimal elevation of hepatic transaminases, this is strong support for the clinical diagnosis of RMSF.
Several serologic methods can establish the diagnosis. A four-fold or greater change in titer between acute and convalescent serum specimens obtained 10-21 days apart is diagnostic when determined by indirect immunofluorescent antibody (IFA) assay. Rickettsia rickettsiae can be identified by immunohistologic staining or PCR of a skin biopsy site. Isolation or PCR assays for detection of the organism in blood are available at the Centers for Disease Control and Prevention (CDC) Reference Laboratory.33
RMSF in its early stages mimics a viral syndrome or "flu" syndrome. As the rash appears and is either macular or maculopapular, the differential diagnosis includes several viral, rickettsial, and bacterial diseases. If the rash becomes petechial, other infectious and non-infectious diseases are considered. (See Table 3.)
| Table 3. Differential Diagnosis for Febrile Illnesses and Centripetally Spreading Rash |
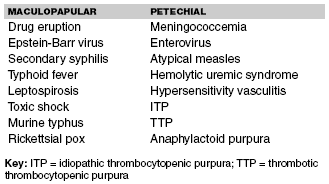 |
Nontoxic patients who present before the end of the first week of the illness have little adverse outcome if they are diagnosed and treated with efficacious antibiotics. Doxycycline, 100 mg every 12 hours, is recommended for children older than age 8. Oral chloramphenicol at 50 mg/kg/day in four divided doses is preferred for younger patients. Inpatient management is warranted for several clinical circumstances, including ill appearance, vital sign changes that include tachycardia with or without hypotension, altered mental status, and poor urine output. Laboratory features that suggest inpatient management include a serum sodium < 130 mEq/liter, thrombocytopenia, or blood-coagulation abnormalities. For inpatients with a clear diagnosis, intravenous chloramphenicol or tetracycline is appropriate. In circumstances in which RMSF cannot be differentiated from meningococcal infection, treatment should include tetracycline or chloramphenicol along with intravenous aqueous penicillin G or parenteral ceftriaxone.
Ulcerative/Pustular
Anthrax. Bacillus anthracis is a gram-positive, aerobic or facultative anaerobic, spore-forming rod that is the causative agent of anthrax. Soil is the primary reservoir for the ubiquitous spore, which can be viable for up to 60 years.34 Cutaneous anthrax is transmitted by exposure to the spores through disrupted skin. Human anthrax is typically transmitted by contact with infected domestic animals such as cattle, sheep, goats, horses, camels, and pigs. Following dermatologic introduction, the incubation period for cutaneous manifestations is 1-10 days.35 Worldwide, 20-100,000 human cases of cutaneous anthrax are estimated to occur annually. The disease is more common in sub-Saharan Africa, Asia, several southern European countries, in the Americas, and in certain areas of Australia.36 The disease was relatively common until the 1940s. Animal vaccination, industrial safety programs, and animal control measures greatly decreased the disease in industrialized nations. Bioterrorism attacks, although rare, are of greater concern for inhalational anthrax.37 The 2001 bioterrorism attacks in the United States infected 22 people; 11 of those cases were cutaneous. With naturally occurring infection, cutaneous anthrax accounts for 95% of the infections.34,35,38 Children account for up to 65% of endemic anthrax cases.39 Pediatric patients between ages 2 and 15 are at highest risk for cutaneous anthrax; both sexes are affected equally. A majority of pediatric patients with cutaneous anthrax will have resolution of the illness without untoward effect. Morbidity results from malignant edema in head and neck infection that may lead to airway compromise. Mortality in untreated cutaneous anthrax may occur in 20% of patients. Mortality is estimated to be from 1-4.2% with appropriate treatment. Mortality results from bacteremic extension to distant foci, including the meninges, or from overwhelming sepsis.39,40
The initial symptoms of cutaneous anthrax are dermatologic. A small, painless but pruritic, erythematous, raised lesion develops at the site of inoculation. Over a period of 1-2 days, there is a progression of a papule to a serous or serosanguinous vesicle 2-3 cm in width. Intensive, non-pitting, non-painful edema evolves within an additional 1-2 days. Occasionally, smaller vesicles ring the initial papule that later enlarge into one large vesicle or bulla. Ultimately, the vesicle ruptures, revealing a well-demarcated, depressed ulcer that is followed by the formation of a painless, black eschar. (See Figure 3.) Within 1-2 weeks, the eschar has sharply-defined margins. Separation of the eschar may take several weeks, and healing occurs with central scarring.41,42
| Figure 3. Painless Black Eschar of Cutaneous Anthrax on an Extremity |
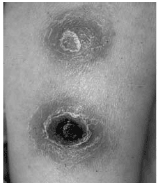 |
Non-dermatologic manifestations are not seen until the localized lesion has progressed to the vesicular stage. Pediatric patients may appear acutely ill, with temperatures of 39-40°C. Regional lymphadenopathy is seen. With head and neck lesions, malignant edema may be associated with respiratory compromise. Altered mental status and progressive neurologic deterioration may occur with patients who experience meningoencephalitis.37
The provisional diagnosis can be bolstered by the presence of gram-positive rods from aspirated culture fluid, exudate from cutaneous lesions, or from cerebrospinal fluid in cases of meningoencephalitis. A definitive diagnosis requires recovery of B. anthracis from a wound, blood, or other sources. The diagnosis may be further confirmed by a four-fold rise in paired sera separated by several weeks to months. Immunoblot, enzyme-linked immunoabsorbent assay for detection of capsular antibody and polymerase chain amplification are available at the CDC and U.S. Armies Medical Research Institute of Infectious Disease, as well as from some state health departments.
The differential diagnosis for a febrile illness, regional lymphadenopathy, and localized vesicobullous or ulcerative skin lesions includes a number of zoonotic diseases. As the appearance of anthrax morphs, the diagnostic considerations change. In the papulovesicular stage, cutaneous anthrax resembles staphylococcal pyoderma and a brown recluse spider bite. A sharply-defined vesicle with a membranous base and surrounding erythema and edema is consistent with cutaneous diphtheria. When similar lesions are multiple (range of 5-100) and widely distributed to include the face, palms, soles, and mucous membranes, rickettsial pox is likely.
Presumptive treatment of ill patients with cutaneous anthrax should include intravenous ciprofloxacin 10 mg/kg every 12 hours. This can be changed to oral therapy at 15 mg/kg as the patient clinically improves. Alternate agents include doxycycline, rifampin, vancomycin, penicillin, chloramphenicol, clindamycin, imipenem and clarithromycin. Any of the latter are suitable if the isolated organism is sensitive.
Vesicular/Blistering
Erythema Multiforme Major. Erythema multiforme minor, erythema multiforme major, and toxic epidermal necrolysis may or may not be related disorders of the skin and mucous membranes. Overlapping features, atypical presentations, and less than standardized terminology has led to confusion in depicting these debilitating and potentially fatal dermatologic conditions. They will be considered variations of the same disease for our discussion. To clarify, in 1866 Ferdinand von Hebra described a dermatologic condition with erythematous macules and flat target lesions unassociated with mucous membrane changes. Today, this clinical entity is uniformly called erythema multiforme minor. In 1922, Stevens and Johnson emphasized mucous membrane involvement in addition to the characteristic rash.43 Stevens-Johnson syndrome (SJS) is synonymous with erythema multiforme major. Most authorities reserve the diagnosis of SJS to cases in which less than 10-20% of the body is involved. When there is erythroderma of greater than 20% of the total body surface area, prominent blistering of large epidermal sheets, and mucous membrane involvement, the term toxic epidermal necrolysis (TEN) is applied. The individuals who use this nomenclature note that SJS and TEN represent escalating expressions of the same entity. However, there are experts who state that there is a significant difference between SJS and TEN in etiology, clinical presentation, and histopathology.44
Neither SJS nor TEN has a well-defined pathogenesis. However, infections and drugs are most often implicated as inciting an immunologic response. In rare circumstances, a previously-healthy patient may develop the illness. In SJS, from 30-50% of patients will have symptoms suggestive of upper respiratory tract infection.45 Specific viral (mumps, polio, herpes simplex, Epstein-Barr virus), bacterial (typhoid, diphtheria, Mycoplasma spp.), and fungal (histoplasmosis, coccidioidomycosis) infections have been documented prior to the onset of SJS. The most common alleged precipitant in SJS is a drug that may have been introduced within the last 7-21 days. Similarly, drugs are most commonly the precipitating factor for TEN. The most frequent drug classes associated with SJS and TEN are antibiotics (penicillins, sulfonamides, cephalosporins), anticonvulsants (phenobarbital, diphenylhydantoin, carbamazepine), nonsteroidal anti-inflammatory drugs (NSAIDs), and anti-retroviral agents.46
All ages can be affected by TEN and SJS. Most often, TEN occurs in the adult population, with a mean age of 45 years in most reports. Most SJS cases occur in late childhood to young adulthood. The mean age in many reports has been around 12 years. There is a slight female prevalence in TEN, whereas there is a slight male predominance in SJS.47
A majority of SJS patients report a prodrome. The symptoms may persist for as little as one day to as long as 1-2 weeks before the onset of skin lesions. The prodrome consists of flu-like symptoms such as malaise, sore throat, nausea, vomiting, myalgia, arthralgia, and fever. Temperature elevations vary from low-grade to more than 40°C.48 Mucositis develops at or shortly after the febrile response. Inflammation of at least two mucous membranes occurs in the course of SJS, with the oral mucosa uniformly involved. Other areas include eyes, vagina, urethra, rectum, throat, tracheobronchial tree, and esophagus.49 Early in the course the patient experiences a burning sensation or pain at the affected mucous membrane. The lips become erythematous and swollen. Bullous lesions rapidly develop. The bullae become friable and ooze reddish-brown-colored or frankly hemorrhagic fluid. Hemorrhagic crusts may extend over the lips onto the palate, esophagus and larynx. Almost one-half of the patients develop eye involvement. Initially the lids get swollen and the bulbar conjunctivae become injected. Conjunctival blisters, corneal blisters, and erosions with subsequent perforation can occur over succeeding days. Crops of painful blisters and erosions with crusting are least common around the vagina and rectum.
The cutaneous manifestations of SJS may precede, occur simultaneously, or follow the mucous membrane lesions.49 The hallmarks of the disease are plaques that develop dusky centers. The patient ultimately exhibits an annular macule referred to as a "target lesion" and areas of erythema that surround a fluid-filled center referred to as an "iris lesion." However, there is significant variation from patient to patient in the morphology, distribution, and time course of the skin changes. Early lesions, which are not pruritic, are erroneously mistaken for hives. Over days isolated, demarcated, erythematous lesions fuse to form larger annular and polycyclic plaques.50 Nonerythematous skin remains intact. Within the erythematous regions, the destruction of epithelial cells from the skin and mucous membranes results in widespread blister formation. Even with slight pressure on the skin, blisters may wrinkle, slide laterally, and separate from the dermis beneath; this is known as Nikolsky's sign. The earliest multiforme rash is symmetric. The trunk and extensor surfaces of the arms are the first involved. Involvement of the legs and dorsa of the hands and feet then occurs. Occasionally, the palms and soles are affected. The perineum and scalp are usually spared. Facial involvement may be minimal to severe. (See Figure 4.)
| Figure 4. Annular, Polycyclic Plaques of SJS with Facial Involvement |
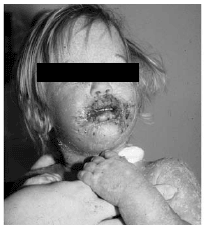 |
The risk of complications in SJS is high. Uniformly, patients have reduced intake and excess fluid losses leading to dehydration. The potential is high for electrolyte balance, protein loss, and anemia. Secondary pyoderma and sepsis are frequent. Frequent complications from ocular involvement include dry eye syndrome, entropion, symblepharon, and corneal damage with permanent visual loss in up to 10% of patients. Uncommon complications include pneumonia, bronchiolitis, acute respiratory distress syndrome, pulmonary hemorrhage, and myocarditis.48,49,51,52
The diagnosis is clinical. There are relatively few febrile disorders with dramatic, polymorphic skin changes. Although there may be some confusion with hypersensitivity drug reaction,53 the presence of target or iris lesions should be discriminatory. When skin blistering is prominent, diagnostic considerations include primary immunobullous dermatoses (pemphigus, pemphigoid, linear IgA dermatosis), bullous contact dermatitis, varicella, vaccinia and herpes simplex. When mucous membrane blistering and ulcerations are prominent, considerations include lichen planus, aphthous stomatitis, inflammatory bowel disease, and lupus erythematosus. When Nikolsky's sign is present, the differential includes staphylococcal scalded skin syndrome (SSSS). Patients with SSSS usually do not look as ill as those with SJS, and mucous membranes are generally spared in SSSS.
As the mortality is in the 1-5% range for SJS associated with < 20% body surface area involvement and in the 30-40% range for > 30% body surface area, hospitalization for observation and supportive care is warranted. The guidelines of symptomatic therapy are similar to that for major burn care and include: intravenous fluid management; skin care with appropriate dressings; nutritional support; and analgesics. Surveillance cultures of skin, blood, and catheters should be taken. Prophylactic antibiotics are not recommended; antibiotics should be reserved for secondary infection or for suspected bacteremia. Antibiotics for suspected or documented infection from Mycoplasma pneumoniae have not been shown to alter outcome. Stopping a suspected drug causing SJS is intuitive, but there are no evidence-based data showing that discontinuation of a drug alters the course of SJS. There is a long-standing controversy about the use of corticosteroids. Corticosteroids at one point were given routinely. However, several retrospective reviews have demonstrated that corticosteroids do not shorten the disease course and they produce more medical complications. Pooled intravenous human globulin has been used but not subjected to randomized trials. Currently, intravenous globulin use is not recommended.54
Serpiginous
Lyme Disease. Lyme disease (LD) results from an infection with the spirochete of the Borrelia spp. The spirochete is transmitted to humans through a bite. Horseflies, mosquitoes, deerflies, and various species of ticks harbor the spirochete. In the United States, the Ixodes spp. of ticks are the main vector. In endemic areas, the infection rate for ticks is high. If an infected tick attaches to the host for more than 36-48 hours, transmission is likely.55 LD is the leading vector-transmitted disease in the United States. The disease has been reported nationwide (with the exception of Montana). Of the cases, 92% are clustered in nine northeastern U.S. states.56 LD has been reported in all age groups, with the highest disease rate by decades of age in the 1-10 year old group. Children and adolescents account for up to 50% of reported cases. In contrast to adults, where there is no gender predilection, in childhood boys are twice as likely as girls to be infected.57
Prodromal symptoms are present in a majority of LD patients. Within 3-30 days of a late summer or autumn bite from an insect harboring the spirochete, patients develop a flu-like illness. Fever and malaise, manifested as observed decreased physical activity or an admission of fatigue, are common. Headache and neck ache or stiffness are prominent, but usually self limiting and resolve in a few days to weeks. Pains in the back, joints, or tendons may occur. These arthralgias and myalgias are localized; tend to affect one or two sites at a time; and are transient, lasting hours to days.58 Ano- rexia, nausea, and vomiting occur in 10% of the patients. Less common systems affected include respiratory (sore throat, conjunctivitis, cervical lymphadenopathy), gastrointestinal (abdominal pain, jaundice),59 and neurologic (altered sleep patterns, nightmares, irritability, hyperactivity).60 These protean prodromal manifestations are then followed by the development of the pathognomonic skin lesion that defines early (stage 1) disease.
Unrecognized and untreated LD leads to further spirochetemia and disseminated illness. The early disseminated disease manifests from 30 to 120 days after inoculation. The common systems affected in early disseminated disease (stage 2) are musculoskeletal, neurologic, cardiac, and ophthalmologic. Among untreated children, approximately 50% develop rheumatologic manifestations. The most common event is pauci-articular arthritis. Weakness secondary to frank myositis has been described.61 Neurologic manifestations occur in 10% of patients with disseminated disease.62 Irrespective of how devastating they are, about 25% of the neurologic symptoms spontaneously remit. A wide range of manifestations occur. They include persistent headaches; insidious aseptic meningitis syndrome; facial pain, paresthesia, numbness and facial droop; vertigo and hearing impairment; and neurocognitive changes including changes in behavior, forgetfulness, and declining school performance.63 About 5% of LD patients develop cardiac involvement. Carditis and heart block are the most common manifestations. Less than 1% develop ophthalmologic abnormalities such as conjunctivitis, keratitis, or optic neuritis.64
If children remain untreated, late disease (stage 3) from four months to one year after inoculation is characterized by rheumatologic complaints. The most common event is pauci-articular arthritis of the large joints, such as the knee, or chronic synovitis of the smaller joints.64
Erythema migrans is the pathognomonic annular, expanding skin lesion of early LD. Erythema migrans occurs in 80-90% of children who develop LD. Coincident with the prodrome, within three days or up to a month after inoculation, a skin lesion appears at the site of a bite. It begins as an asymptomatic, erythematous, flat macule or red papule. Over 24-48 hours, the macule or papule expands to become a mildly stinging, mildly pruritic, round or oval lesion of about 5 cm in diameter. The perimeter of the lesion is typically bright red and warm to the touch. The central area generally clears. (See Figure 5.) Over subsequent days to a week, the lesion advances. The median diameter reaches to 16 cm, but extension may reach 68 cm.65 If unrecognized or untreated, the primary lesion fades over weeks to a year (mean is 28 days). As the lesion fades, the skin has a bluish hue. While the primary lesion is still present, up to one-half of the patients develop multiple, satellite lesions in proximity to the original lesion. These metastatic lesions are annular, flat and erythematous, but they are smaller and less expansive. Secondary skin lesions also fade over days to subsequent weeks.64
| Figure 5. Advancing Annular Eruption of Erythema Migrans on an Extremity |
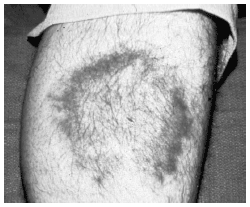 |
It is difficult to confirm LD during an ED encounter. The spirochetes are present in the erythema migrans eruption. However, a biopsy specimen of the perimeter of the skin lesion yields a spirochete only in special media that is not commercially available. Dark field visualization of the spirochete is not possible in the emergency setting. Serology is the established technique for laboratory diagnosis. The emergency physician who submits a blood sample for enzyme-linked immunosorbent assay for anti-B. burgdorferi IgM and IgG antibodies in a patient with erythema migrans 3-30 days after inoculation will find no serologic response.66 In rare circumstances, B. burgdorferi may be isolated from a blood culture in a child with solitary erythema migrans.67
The non-dermatologic prodromal features are consistent with a flu-like syndrome. As LD occurs between April and October, with 50% of cases in June and July, the diagnostic differential considerations are rickettsial illnesses and enteroviral illnesses. When the characteristic rash occurs along with the prodrome, there should be little difficulty with the etiology.
The patient with early localized disease may be successfully treated with a 14-21 day therapeutic regimen. Doxycycline, 100 mg twice a day, is the drug of choice for children ages 8 and older. For children younger than age 8, amoxicillin, 50 mg/kg/day divided into 3 doses, is recommended. For patients allergic to penicillins, an alternate drug is cefuroxime 30 mg/kg/day in 2 divided doses.68 With successful treatment, many of the nonspecific signs and symptoms of LD may persist for several weeks. The erythema migrans rash resolves within several days of initiation of treatment. Of great importance, treatment of early localized disease prevents development of later stages of LD.
Mixed
Kawasaki Disease. Kawasaki disease (KD) is a vasculitis process of unknown etiology. An infectious cause is suggested by four epidemiologic features: 1) The syndrome appears in epidemic cycles with periodicity of 2-3 years; 2) There is seasonality, with occurrence primarily in late winter and early spring; 3) There are often clusters of cases within a geographic locale; and 4) The syndrome may appear in siblings in a household with simultaneous or near-simultaneous onset of symptoms.69 No parasitic or rickettsial organisms have been recovered in patients with the syndrome. No single virus has been consistently cultured or serologically confirmed to be associated with KD. Group A beta-hemolytic Streptococcus can occasionally be cultured from the patient's pharynx, and isolated cases of bacteremia have been reported during KD. However, there is no evidence that the bacteria recovered have been a stimulus for the development of the disease.70,71 Irrespective of the origin of the syndrome, immunoregulatory abnormalities are prominent in affected patients. Consistently there is an increased production of interleukins 1, 2, 6 and 8; tumor necrosis factor alpha; and the appearance of circulating antibodies that are cytotoxic to cytokine-activated endothelial cells.72 The findings of serial immunoregulatory abnormalities has led many authorities to speculate that a patient may be programmed to respond clinically and immunologically to an as yet ill-defined infectious pathogen.73
KD has been described worldwide in children of all racial groups. Within the United States, rates of occurrence are highest in children of Asian and Pacific descent; intermediate in African Americans and Hispanics; and lowest in whites.74 KD has been diagnosed in patients as young a two months and as old as 19 years. About 80% of the patients are younger than age 5 and 50% are younger than age 2. The peak incidence in the majority of studies has been between ages 1-2. There is a 1.5:1 male to female predominance of KD. Fatal KD (0.4-1.7% of affected children) is more common among males and in children younger than age 2 and older than age 8.75 Fatalities are typically related to cardiac pathology and are rare within the first 10 days of the illness but can result from myocarditis, pericarditis, endocarditis, and inflammation of the conduction system. Seventy percent of mortalities occur during the subacute phase (day 11-50) as a result of coronary artery abnormalities. Mortality results from acute myocardial infarction, coronary aneurysmal dilatation, thrombosis, stenosis, or rupture.76
There is a variation in the non-dermatologic presentation of KD; however, fever is a constant feature. In the majority of cases, the initial manifestation of KD is fever that remains elevated for 6-24 days.77 Beside fever, eye findings are prominent diagnostic features. A red eye has been reported in 89-90% of patients. Bilateral conjunctival injection has its onset one day before or concurrent with the onset of the fever. This conjunctival injection is usually more prominent in the bulbar conjunctiva than in the tarsal or palpebral conjunctiva.78 Engorged vessels originate at the canthi and course toward, but generally do not reach, the limbus. The red eye is generally unassociated with exudate or crusting of the eyelids.
Beyond the eye findings, classic patients develop two of the following three nondermatologic features in the acute phase (days 1-5).79 These include changes in the lips and oral cavity; peripheral extremities; and cervical architecture. The oral changes are heralded by a sore throat. The pharyngeal mucosa is injected without exudate or petechiae. The tongue surface resembles a strawberry. The uvula or epiglottis may be inflamed. The lips are erythematous, dry, and cracked. The hand and foot changes are the most helpful diagnostic features as they are the most specific. The hands and feet become erythematous and swollen. The edema ranges from subtle to obvious. The patient may have an inability to make a fist, seem uncomfortable with standing, or refuse to walk. The least frequent principle feature is cervical alteration, which occurs in less than one-half of patients. In descending order they include unilateral lymphadenopathy (at least 1.5 cm diameter), bilateral cervical lymphadenopathy, and suppurative bilateral lymphadenitis.80
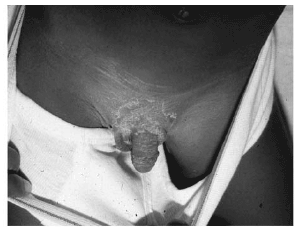
The rash of KD appears within the first 2-7 days of the illness. The rash is considered mixed or polymorphous, due to its variable appearance. The most common is an erythematous eruption that is maculopapular and resembles scarlet fever (scarlatiniform) or measles (morbilliform). Less common are erythematous plaques or papules. The latter may simulate erythema multiforme. (See Figure 6.) Other variations include urticarial, erythema marginatum-like, vesiculopustular, or petechial.81 The rash usually involves the trunk and extremities, but on occasion may be especially noticeable or restricted to the perineal area.82 The perineal rash is dry and tends to desquamate in the first few days of the illness. (See Figure 7.) This desquamation differs from the late periungual desquamation, transverse furrows of fingernails (Beau's lines), and nail loss that occurs in the subacute phase (week 1-3).
| Figure 6. Generalized Erythema Multiforme-like Eruption in Kawasaki Disease |
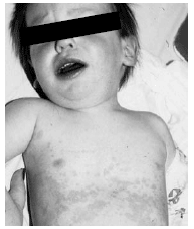 |
| Figure 7. Perineal Desquamation in Early Stage of Kawasaki Disease |
 |
Laboratory abnormalities in KD are nonspecific. Analysis of effusions, cerebrospinal fluid, and blood reflect a widespread release of inflammatory mediators. Total white blood cell counts are elevated to values of 20,000/mm3 or greater in more than one-half the patients during the acute febrile phase of the illness. There is a predominance of polymorphonuclear leukocytes with both mature and band forms. The C-reactive protein is higher in those patients who present within the first ten days of the illness.83 Erythrocyte sedimentation rates rise more slowly in response to the inflammatory stimulus. Platelet counts have a similar time course. The platelet count is normal during the early acute febrile phase of the illness. By the end of the first week elevations occur, and peaks are noted between the second and third weeks of the illness. Values generally ascend to greater than 1,000,000 platelets/mm3. Rarely, thrombocytopenia may be observed within the early phase of the illness, with platelet counts returning to normal within 1-2 weeks.84 Other hematologic features include a normocytic, normochromic anemia. Measurement of liver enzymes occasionally reveals elevated transaminase levels or mild hyperbilirubinemia. Urinalysis commonly reveals a sterile pyuria. An electrocardiogram within the first week of the illness is often remarkable for tachycardia and nonspecific P-R prolongation or flattening of the T waves. It generally takes longer than a 7-10 day timeframe to develop electrocardiographic changes reflecting inflammation of the AV conduction system; microvascular angitis of the coronary arteries; and pericardial, myocardial, or endocardial inflammation.
Classic KD is diagnosed when the patient has fever more than 5 days, along with four of following five criteria: conjunctivitis, mucositis, extremity changes, rash, or abnormalities of cervical architecture. Approximately 7-10% of patients with KD have incomplete presentations; as few as 1-2 diagnostic features are present along with compatible laboratory findings.85 With incomplete KD, diagnostic uncertainty is increased. Further, confusion occurs and the diagnosis of KD is overlooked if the patient with a diffuse vasculitis presents with non-cardiovascular organ system involvement. Neurologic, gastrointestinal, musculoskeletal, genitourinary, and pulmonary systems may be involved with KD.
Salicylates are administered from as low as 30-50 mg/kg/day to as high as 80-100 mg/kg/day in the acute phase of the illness. Intravenous gammaglobulin was first administered at 400 mg/kg for 5 days in uncontrolled trials. The current recommendation is to administer 2 gm/kg as a single infusion. Attempts should be made to begin gammaglobulin therapy before the seventh day of the illness to provide maximum protection against development of coronary abnormalities.86,87 Earlier studies with corticosteroid therapy in children with KD showed an increased risk of coronary aneurysms. Recent studies have shown that corticosteroids resulted in faster resolution of fever, more rapid improvement in inflammatory markers, and shorter length of hospitalization.88 Multicenter, double-blind, placebo-controlled studies with long-term outcomes are necessary before corticosteroids can be recommended.
Conclusion
The early identification of rashes that may herald critical diseases is an essential skill for the ED physician. Prompt diagnosis and early implementation of effective therapies will decrease the morbidity and mortality associated with these critical illnesses.
References
1. Nelson DS, Walsh K, Fleisher GR. Spectrum and frequency of pediatric illness presenting to a general community hospital emergency department. Pediatrics 1992;90:5-10.
2. Krauss BS, Harakal T, Fleisher GR. The spectrum and frequency of illness presenting to a pediatric emergency department. Pediatr Emerg Care 1991;7:67-71.
3. Shivaram V, Christoph RA, Hayden GF. Skin disorders encountered in a pediatric emergency department. Pediatr Emerg Care 1993;9:202-204.
4. Lynch PJ, Edminster SC. Dermatology for the nondermatologist: a problem-oriented system. Ann Emerg Med 1984;13:603-606.
5. Leggiadro RJ. Systemic meningococcal infection and complement deficiency. Pediatr Infect Dis J 2003;22:760-761.
6. Dashefsky B, Teele DW, Klein JO. Unsuspected meningococcemia. J Pediatr 1983;102:69-72.
7. Stovall SH, Schutze GE. Meningococcal infections in children from Arkansas. Pediatr Infect Dis J 2002;21:366-370.
8. Wong VK, Hitchcock W, Mason WH. Meningococcal infections in children: a review of 100 cases. Pediatr Infect Dis J 1989;8:224-227.
9. Stiehm ER, Damrosch DS. Factors in the prognosis of meningococcal infection. Review of 63 cases with emphasis on recognition and management of the severely ill patient. J Pediatr 1966;68:457-467.
10. Werne CS. Gastrointestinal disease associated with meningococcemia. Ann Emerg Med 1984;13:471-473.
11. Kaufman B, Levy H, Zaleznak BD, et al. Statistical analysis of 242 cases of meningococcus meningitis. J Pediatr 1951;38:705-716.
12. Marzouk O, Thomson AP, Sills JA, et al. Features and outcome in meningococcal disease presenting with maculopapular rash. Arch Dis Child 1991;66:485-487.
13. Toews WH, Bass JW. Skin manifestations of meningococcal infection; an immediate indicator of prognosis. Am J Dis Child 1974;127:173-176.
14. Weiss PJ, Frum DK, Kennedy CA. Large erythematous patch as the presenting manifestation of meningococcemia. Cutis 1992;50:417-418.
15. van Deuren M, van Dijke BJ, Koopman RJ, et al. Rapid diagnosis of acute meningococcal infections by needle aspiration or biopsy of skin lesions. BMJ 1993;306:1229-1232.
16. Boyer D, Gordon RC, Baker T. Lack of clinical usefulness of a positive latex agglutination test for Neisseria meningitidis/Escherichia coli antigens in the urine. Pediatr Infect Dis J 1993;12:779-780.
17. Wright MS. Purpura in childhood. Pediatr Emerg Med Reports 2000;5:1-10.
18. Varon J, Chen K, Sternbach GL. Rupert Waterhouse and Carl Friderichsen: adrenal apoplexy. J Emerg Med 1998;16:643-647.
19. Jacobs RF, Hsi S, Wilson CB, et al. Apparent meningococcemia: clinical features of disease due to Haemophilus influenzae and Neisseria meningitidis. Pediatrics 1983;72:469-472.
20. Saks MA, Karras D. Emergency medicine and the public's health: emerging infectious diseases. Emerg Med Clin North Am 2006;24:1019-1033.
21. Dhodapkar K, Corbacioglu S, Chang MW, et al. Purpura fulminans caused by group A beta-hemolytic Streptococcus sepsis. J Pediatr 2000;137:562-567.
22. Kuppermann N, Inkelis SH, Saladino R. The role of heparin in the prevention of extremity and digit necrosis in meningococcal purpura fulminans. Pediatr Infect Dis J 1994;13:867-873.
23. Krzelj V, Petri NM, Mestrovic J, et al. Purpura fulminans successfully treated with hyperbaric oxygen—a report of 2 cases. Pediatr Emerg Care 2005;21:31-34.
24. De Kleijn ED, Joosten KF, Van Rijn B, et al. Low serum cortisol in combination with high adrenocorticotrophic hormone concentrations are associated with poor outcome in children with severe meningococcal disease. Pediatr Infect Dis J 2002;21:330-336.
25. Fischer M, Hilinski J, Stephens DS. Adjuvant therapy for meningococcal sepsis. Pediatr Infect Dis J 2005;24:177-178.
26. Paddock CD, Holman RC, Krebs JW, et al. Assessing the magnitude of fatal Rocky Mountain spotted fever in the United States: comparison of two national data sources. Am J Trop Med Hyg 2002;67:349-354.
27. Treadwell T, Holman R, Clarke M, et al. Rocky Mountain spotted fever in the United States, 1993-1996. Am J Trop Med Hyg 2000;63:21-26.
28. Sexton DJ, Kaye KS. Rocky Mountain spotted fever. Emerg Med Clin North Am 2002;86:351-360, vii-viii.
29. Lowenstein R. Deadly viral syndrome mimics. Emerg Med Clin North Am 2004;22:1051-1065, ix-x.
30. Walker DH, Henderson FW, Hutchins GM. Rocky Mountain spotted fever: mimicry of appendicitis or acute surgical abdomen? Am J Dis Child 1986;140:742-744.
31. Griffith GL, Luce EA. Massive skin necrosis in Rocky Mountain spotted fever. South Med J 1978;71:1337-1340.
32. Rubio T, Riley HD Jr., Nida JR, et al. Thrombocytopenia in Rocky Mountain spotted fever. Am J Dis Child 1968;116:88-96.
33. Singh-Behl D, La Rosa SP, Tomecki KJ. Tick-borne infections. Dermatol Clin 2003;21:237-244, v.
34. Tutrone WD, Scheinfeld NS, Weinberg JM. Cutaneous anthrax: a concise review. Cutis 2002;69:27-33.
35. Vijaikumar M, Thappa DM, Karthikeyan K. Cutaneous anthrax: an endemic outbreak in south India. J Trop Pediatr 2002;48:225-226.
36. Organization WH. Epidemic and Pandemic Alert and Response (EPR), Anthrax: World Health Organization, 2006.
37. Holty JE, Kim RY, Bravata DM. Anthrax: a systematic review of atypical presentations. Ann Emerg Med 2006;48:200-211.
38. Maguina C, Flores Del Pozo J, Terashima A, et al. Cutaneous anthrax in Lima, Peru: retrospective analysis of 71 cases, including four with a meningoencephalic complication. Rev Inst Med Trop Sao Paulo 2005;47:25-30.
39. Kaya A, Tasyaran MA, Erol S, et al. Anthrax in adults and children: a review of 132 cases in Turkey. Eur J Clin Microbiol Infect Dis 2002;21:258-261.
40. Lanska DJ. Anthrax meningoencephalitis. Neurology 2002;59:327-334.
41. Ciftci E, Ince E, Dogru U. Traditions, anthrax, and children. Pediatr Dermatol 2002;19:36-38.
42. Meade J. Images in emergency medicine. Cutaneous anthrax. Ann Emerg Med 2004;43:664, 673.
43. Stevens AM, Johnson FC. A new eruptive fever associated with stomatitis and ophthalmia: report of two cases in children. Am J Dis Child 1922;24:526.
44. Leaute-Labreze C, Lamireau T, Chawki D, et al. Diagnosis, classification, and management of erythema multiforme and Stevens-Johnson syndrome. Arch Dis Child 2000;83:347-352.
45. Levy M, Shear NH. Mycoplasma pneumoniae infections and Stevens-Johnson syndrome. Report of eight cases and review of the literature. Clin Pediatr (Phila) 1991;30:42-49.
46. Rasmussen JE. Erythema multiforme. Should anyone care about the standards of care? Arch Dermatol 1995;131:726-729.
47. Stewart MG, Duncan NO 3rd, Franklin DJ, et al. Head and neck manifestations of erythema multiforme in children. Otolaryngol Head Neck Surg 1994;111:236-242.
48. Prendiville JS, Hebert AA, Greenwald MJ, et al. Management of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. J Pediatr 1989;115:881-887.
49. Araujo OE, Flowers FP. Stevens-Johnson syndrome. J Emerg Med 1984;2:129-135.
50. Cohen B. The many faces of erythema multiforme. Contemp Pediatr 1994;11:19-39.
51. Rottermann EM, Julia MV, Rovira J, et al. Esophageal stenosis following Stevens-Johnson syndrome. Treatment with balloon dilation. Clin Pediatr (Phila) 1990;29:336-338.
52. Edell DS, Davidson JJ, Muelenaer AA, et al. Unusual manifestation of Stevens-Johnson syndrome involving the respiratory and gastrointestinal tract. Pediatrics 1992;89:429-432.
53. Bachot N, Roujeau JC. Physiopathology and treatment of severe drug eruptions. Curr Opin Allergy Clin Immunol 2001;1:293-298.
54. Bachot N, Roujeau JC. Intravenous immunoglobulins in the treatment of severe drug eruptions. Curr Opin Allergy Clin Immunol 2003;3:269-274.
55. Piesman J, Maupin GO, Campos EG, et al. Duration of adult female Ixodes dammini attachment and transmission of Borrelia burgdorferi, with description of a needle aspiration isolation method. J Infect Dis 1991;163:895-897.
56. Baumgarten JM, Montiel NJ, Sinha AA. Lyme disease—part I: epidemiology and etiology. Cutis 2002;69:349-352.
57. Williams CL, Strobino B, Lee A, et al. Lyme disease in childhood: clinical and epidemiologic features of ninety cases. Pediatr Infect Dis J 1990;9:10-14.
58. Belman AL. Neurologic complications of Lyme disease in children, experience in an endemic area and review of the literature. Int'l Pediatr 1992;7:136-142.
59. Edwards KS, Kanengiser S, Li KI, et al. Lyme disease presenting as hepatitis and jaundice in a child. Pediatr Infect Dis J 1990;9:592-593.
60. Christy C, Siegel DM. Lyme disease — what is it?, what it isn't? Contemp Pediatr 1995;12:64-84.
61. Reimers CD, de Koning J, Neubert U, et al. Borrelia burgdorferi myositis: report of eight patients. J Neurol 1993;240:278-283.
62. Coyle PK, Schutzer SE. Neurologic presentations in Lyme disease. Hosp Pract (Off Ed) 1991;26:55-66; discussion 66, 69-70.
63. Bingham PM, Galetta SL, Athreya B, et al. Neurologic manifestations in children with Lyme disease. Pediatrics 1995;96:1053-1056.
64. Montiel NJ, Baumgarten JM, Sinha AA. Lyme disease—part II: clinical features and treatment. Cutis 2002;69:443-448.
65. Bruhn FW. Lyme disease. Am J Dis Child 1984;138:467-470.
66. Shapiro E, Gerber MA. Lyme disease. Clin Infect Dis 2000;31:533-542.
67. Arnez M, Ruzic-Sabljic E, Ahcan J, et al. Isolation of Borrelia burgdorferi sensu lato from blood of children with solitary erythema migrans. Pediatr Infect Dis J 2001;20:251-255.
68. Eppes SC, Childs JA. Comparative study of cefuroxime axetil versus amoxicillin in children with early Lyme disease. Pediatrics 2002;109:1173-1177.
69. Fink HW. Kawasaki syndrome in twins. Pediatr Infect Dis 1984;3:372-373.
70. Johnson D, Azimi P. Kawasaki disease associated with Klebsiella pneumoniae bacteremia and parainfluenza type 3 virus infection. Pediatr Infect Dis 1985;4:100.
71. Cox F, Foshee W, Miller J, Jr., et al. Simultaneous Kawasaki disease and group A streptococcal pharyngitis. Clin Pediatr (Phila) 1993;32:48-50; discussion 51-42.
72. Lin CY, Lin CC, Hwang B, et al. Serial changes of serum interleukin-6, interleukin-8, and tumor necrosis factor alpha among patients with Kawasaki disease. J Pediatr 1992;121:924-926.
73. Brosius C, Newburger J, Burns J, et al. Increased prevalence of atopic dermatitis in Kawasaki disease. Pediatr Infect Dis J 1988;7:863-866.
74. Holman R, Curns A, Belay E, et al. Kawasaki syndrome hospitalizations in the United States, 1997 and 2000. Pediatrics 2003;112:495-501.
75. Stockheim JA, Innocentini N, Shulman ST. Kawasaki disease in older children and adolescents. J Pediatr 2000;137:250-252.
76. Hamaoka K, Kamiya Y, Sakata K, et al. [Evaluation of coronary hemodynamics and coronary reserve in children with coronary sequelae of Kawasaki disease]. J Cardiol 1991;21:423-435.
77. Levy M, Koren G. Atypical Kawasaki disease: analysis of clinical presentation and diagnostic clues. Pediatr Infect Dis J 1990;9:122-126.
78. Smith LB, Newburger JW, Burns JC. Kawasaki syndrome and the eye. Pediatr Infect Dis J 1989;8:116-118.
79. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics 2004;114:1708-1733.
80. Waggoner-Fountain LA, Hayden GF, Hendley JO. Kawasaki syndrome masquerading as bacterial lymphadenitis. Clin Pediatr 1995;34:185-189.
81. Yamamoto LG, Martin JE. Kawasaki syndrome in the ED. Am J Emerg Med 1994;12:178-182.
82. Krowchuk DP, Bass J, Elgart GW. Kawasaki disease with an exanthem limited to the diaper area. Am J Dis Child 1988;142:1136-1137.
83. Anderson MS, Burns J, Treadwell TA, et al. Erythrocyte sedimentation rate and C-reactive protein discrepancy and high prevalence of coronary artery abnormalities in Kawasaki disease. Pediatr Infect Dis J 2001;20:698-702.
84. Venglarcik JS 3rd, Ayas M, Woods T. Severe thrombocytopenia as a presenting manifestation of Kawasaki disease. Arch Pediatr Adolesc Med 1995;149:215-217.
85. Barone SR, Pontrelli LR, Krilov LR. The differentiation of classic Kawasaki disease, atypical Kawasaki disease, and acute adenoviral infection: use of clinical features and a rapid direct fluorescent antigen test. Arch Pediatr Adolesc Med 2000;154:453-456.
86. Terai M, Shulman ST. Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose. J Pediatr 1997;131:888-893.
87. Tse SM, Silverman ED, McCrindle BW, et al. Early treatment with intravenous immunoglobulin in patients with Kawasaki disease. J Pediatr 2002;140:450-455.
88. Sundel RP, Baker AL, Fulton DR, et al. Corticosteroids in the initial treatment of Kawasaki disease: report of a randomized trial. J Pediatr 2003;142:611-616.
Rashes are common in the emergency department and may be a challenge diagnostically. The authors review rashes that the clinician cannot afford to miss.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.