Acute Disseminated Encephalomyelitis: Pediatric Multiple Sclerosis or a Distinct Entity?
Acute Disseminated Encephalomyelitis: Pediatric Multiple Sclerosis or a Distinct Entity?
Authors: Roytesa R. Savage, MD, Assistant Professor of Pediatrics, Brody School of Medicine at East Carolina University, Greenville, North Carolina; Ronald M. Perkin, MD, MA, Professor and Chairman, Department of Pediatrics, Brody School of Medicine at East Carolina University; Medical Director, Children's Hospital of Eastern North Carolina, Greenville, North Carolina.
Peer Reviewer: Grant S. Lipman, MD, Clinical Instructor of Surgery, Department of Emergency Medicine, Stanford University School of Medicine.
Introduction
Defined as an inflammatory demyelinating disease process that affects the brain and spinal cord, acute disseminated encephalomyelitis (ADEM) has been called the most common white matter disease in children.1 The presentations can vary symptomatically as well as radiographically. The patient history may include a recent viral infection or vaccination; historically, it has been known as postinfectious or postvaccination ADEM. Post infectious encephalomyelitis appears to be decreasing in incidence with the eradication of smallpox and the vaccination for measles. Mycoplasma is commonly implicated as a bacterial cause of ADEM but other viruses and vaccinations have been implicated as well. In most modern series, however, the majority of cases present after having non-specific symptoms of a respiratory or gastrointestinal illness that is never attributed to a specific pathogen.2
As its name implies, ADEM is typically a single, acute episode of demyelination that can simultaneously affect multiple areas of the central nervous system (CNS), including the optic nerves, brain, brainstem, and spinal cord.2-7 Like other inflammatory demyelinating diseases of the CNS, such as acute transverse myelitis (ATM), it most often develops 1-4 weeks after a systemic illness or vaccination and is believed to be triggered by the collateral activation of an immune response directed against self-antigens in myelin.2 Most studies find no clear gender predominance.2,6
ADEM is a polysymptomatic (multifocal neurologic signs and deficits) demyelinating disorder associated with encephalopathy.7 There is a wide variation in the clinical presentation and severity of ADEM; the neurologic features are dependent upon the location of the actual lesions within the brain or CNS. Some of the typical symptoms other than encephalopathy include headache, vomiting, malaise, and fever. The neurologic signs include ataxia, tremors, cranial nerve palsies, seizures, and visual loss due to optic neuritis. There are no specific biological markers for ADEM; therefore, the diagnosis is based on clinical and radiologic features.
MRI has become a major part of the diagnosis and management of patients with ADEM.3
First-line treatment for ADEM includes supportive care and steroids. The length of therapy generally depends on the patient's severity upon presentation. Many of these children will require ICU care but the overall prognosis is good.
While ADEM is typically monophasic, recurrences can occur; this raises the possibility of a diagnosis of multiple sclerosis (MS).3,6,7 MS is uncommon in adolescents and even rarer in pre-pubescent children, but there has been increased recognition of pediatric MS over the past two decades.7-10 This is, in part, due to the advent of MRI, which has established sensitive detection of white matter abnormalities.
There are limited data about the risk of progression to MS after an initial demyelinating event in childhood. Identifying MS in childhood is important for the overall management of both physical and quality-of-life issues.
This review summarizes current knowledge of the main aspects of ADEM and pediatric MS.
Long-Term Outcome
Source: Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology 2002;59:1224-1231.
The clinical and neuroimaging findings and outcomes of 84 consecutive children with ADEM seen between March 1988 and July 2000 were followed in a prospective study at the National Pediatric Hospital, Buenos Aires, Argentina. The patients' mean age at onset of ADEM was 5.3 ± 3.9 years (range, 4-16 years); males outnumbered females by 1.8:1. A febrile infectious illness or vaccination preceded the onset of neurologic symptoms by 12.1 days (range, 2-30 days) in 74% of patients. No prodrome was recognized in 26% of cases. The most frequent presenting signs were hyperreflexia (85%), acute hemiparesis (75%), mental changes (69%), and ataxia (50%). MRI abnormalities consisted of small (62%) or large (24%) areas of cerebral demyelination, bithalamic lesions (12%), and acute hemorrhagic encephalomyelitis (2%). Cerebral spinal fluid (CSF) was abnormal in 28% of those with lymphocytic pleocytosis or mildly elevated protein; none showed oligoclonal bands. Complete or almost complete recovery and resolution of MRI lesions occurred between 3 and 24 months after steroid treatment (mean, 7.2 months). At mean follow-up of 6.6 ± 3.8 years (range, 1 to 19 years), 90% had no further relapse and 10% had 1 relapse, between 2 months and 8 years (mean, 2.9 years) after the first episode. Disability was absent to mild in 89% and moderate to severe in 11% of patients. Disability was not related to initial MRI findings, but was related to optic nerve involvement in 19 patients. Residual deficits included hemiparesis (8%), partial epilepsy (6%), visual impairment (6%), and mental handicap (4%).
Commentary
This study supports the following statements: Childhood ADEM is an acute inflammatory demyelinating disease that is typically preceded by a viral febrile illness or vaccination; affects boys more frequently than girls; and usually follows a monophasic course, with full recovery in 89% of patients. Although the study demonstrated that 10% of patients with ADEM have a biphasic course, the authors argue that even in relapsing cases, the distinction between ADEM and MS was possible on the basis of long-term clinical and neuroimaging follow-up and the absence of oligoclonal bands in CSF.
More on Prognosis
Source: Mikaeloff Y, Suissa S, Vallee L, et al. First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr 2004;144:246-252.
This study, the KIDMUS pediatric MS cohort, derived from the European Database for Multiple Sclerosis (EDMUS),11 tracked 296 patients younger than age 16 after an initial demyelinating episode (ADEM, MS, ATM, optic neuritis). In this study, 168 patients (57%) experienced two or more episodes of demyelination and were diagnosed with MS. Of the 296 patients, 40% initially presented with ADEM, defined by KIDMUS investigators as altered mental status with polysymptomatic presentation and MRI "suggestive of ADEM" with poorly limited lesions and involvement of the thalamus or basal ganglia. Of the 119 children with an initial diagnosis of ADEM, 29% experienced recurrent demyelination and were re-classified as having MS. Unfortunately, details about the subsequent demyelination episodes are limited, so it is impossible to determine which of these cases may have actually represented recurrence of multiphasic ADEM.7,10,12
Predictors for a second attack in the KIDMUS study included optic neuritis, age older than 10, or an MRI "suggestive of MS" with multiple well-defined periventricular or subcortical lesions. A decreased risk of MS was found in patients who presented with myelitis or altered mental status.
Commentary
This represents the largest prospective series of pediatric demyelinating disease to date. The finding that older patients and patients with relapsing episodes of demyelination are more likely to go on to have MS and significant disabilities is similar to that reported in a comparison of 28 patients with ADEM versus 13 patients with MS.10 However, both studies are limited by relatively short follow-up (3-6 years). More population-based, longitudinal studies are essential to determine which pediatric patients are at highest risk for MS after an initial demyelinating event so that we can identify those who may benefit from early initiation of disease modifying therapy.
Indications for Early, Aggressive Therapy
Source: Khurana DS, Melvin JJ, Kothare SV, et al. Acute disseminated encephalomyelitis in children: discordant neurologic and neuroimaging abnormalities and response to plasmapheresis. Pediatrics 2005;116:431-436.
This is a retrospective record review of 13 children admitted to a single hospital with a diagnosis of ADEM during a six-year period (1998-2003). The diagnosis of ADEM was established by clinical signs and symptoms, CSF changes, and multifocal involvement of deep gray and white matter based on MRI. Patients ranged in age from 5 to 17 years; there were 5 girls and 8 boys.
Although all of the children developed MRI changes in subcortical white matter and/or deep gray matter, brainstem, cerebellum, and spinal cord, seven children had initial MRIs that were normal even on retrospective review. The time lag before lesions could be identified on MRI ranged from 2 days to 25 days after the onset of neurologic symptoms. Five of the seven patients with initially normal MRI findings had a rapid progressive course. Six of 13 children had rapid progression of symptoms that required prolonged care in the ICU. All six of these children had lesions in the brain stem. The six patients who had a rapidly progressive course were given intravenous immunoglobulin (IVIG) in addition to corticosteroids. When they failed to show significant improvement, all six had plasmapheresis.
Duration of follow-up ranged from 1.5 to 5 years (median: 2 years). Eight of 13 children had complete recovery. Three children had a residual spastic paraparesis; one child had persistent ophthalmoplegia and cranial nerve dysfunction; and one child was lost to follow-up. One child developed new symptoms and new lesions on MRI within six months of initial presentation. She received another course of methylprednisolone with clinical improvement and resolution of MRI abnormalities. She did not develop any additional episodes of demyelination in a five-year period.
All the patients who received plasmapheresis had a prolonged ICU stay and were discharged to inpatient rehabilitation. At the time of discharge, all had improved. At the time of the last follow-up, one had an excellent recovery with no deficits; three children had residual spastic paraparesis (expanded disability status scale, EDSS, 5-6.5), and one child had persistent ophthalmoplegia and cranial nerve dysfunction (EDSS, 3.5). One child was lost to follow-up.
Commentary
This small study provides clinically useful information. First, it describes a subset of children who have a more aggressive course. These children have extensive lesions in the brainstem or cervical cord; they are more likely to require ICU care, have poor response to corticosteroids, and have long-term disability. Second, although MRI is regarded as the imaging modality of choice in diagnosing ADEM, a significant proportion (7 of 13) of patients in this study had a normal MRI at a time of maximal neurologic deficits. Some did not exhibit significant imaging changes until several days into the illness. A lag between clinical course and imaging changes has been described in adults,13 but this is the first report in children. This points to the heterogeneous nature of ADEM and its varied clinical presentation. It also suggests that treatment should not be delayed when clinical suspicion is high. Finally, on the basis of their experiences, the authors suggest that plasmapheresis should be considered as a treatment option for patients with ADEM, especially when the course is aggressive or severe disease has not responded to corticosteroids and IVIG. However, immunosuppressive or immunomodulatory therapies for ADEM are used on an empirical basis, without the benefit of prospective research data.2 A multicenter prospective study to address the outcome of the use of different therapeutic modalities in the treatment of ADEM is warranted.
MS or ADEM?
Source: Banwell B, Shroff M, Ness JM, Jeffrey D, et al. MRI features of pediatric multiple sclerosis. Neurology 2007;68(Suppl 2):S46-S53.
This review summarized the available literature on MRI in pediatric MS, outlined the specific features of other disorders affecting the CNS white matter in children, compared the MRI appearance of MS in children to seminal neuroimaging studies in adult-onset MS, and discussed the potential role of advanced MRI technologies delineating the underlying pathobiology of acquired demyelinating disease in children.
Although the MRI features of MS in children have similarity to adult-onset MS, children tend to have fewer lesions and a lower propensity for lesions to enhance with gadolinium. The MRI findings in children presenting with a clinical phenotype of ADEM may be indistinguishable for the first attack of MS.
Commentary
MRI criteria specific for pediatric-onset MS and criteria predictive of MS outcome in children experiencing a first demyelinating event will be challenged by the overlap in MRI features between acute monophasic demyelinating syndromes and MS, particularly in younger children. Emergence of new clinically silent lesions on MRI scans separated by at least three months is characteristic of MS. Newer MRI techniques evaluating white matter biochemistry and integrity in the youngest MS patients may provide new insights into the relative contributions of inflammation and neurodegeneration in MS.
Improvement with Steroids
Source: Gupte G, Stonehouse M, Wassmer E, et al. Acute disseminated encephalomyelitis: a review of 18 cases in childhood. J Paediatr Child Health 2003;39:336-342.
This retrospective review of the charts of 18 children with a diagnosis of ADEM established in a tertiary referral center from 1995-2000 was performed with particular attention to clinical features, investigations, and treatment. The mean age of the 18 children at presentation was 8 years (range, 3.5 months to 17 years). The most common presenting features were ataxia (10 cases), followed by headache (8 cases), and weakness (5 cases). A brain CT scan was performed in 11 children, but was abnormal in only 2 cases. CSF examination was performed in 13 children; 8 children had normal CSF examination, oligoclonal bands were detected in 4 children, and 1 child had a CSF lymphocytosis. All children had MRI scans, and the commonest abnormality found was of high signed on T2-weighted images in the subcortical weight matter of the hemispheres, basal ganglia, and brainstem. Seven children had abnormalities in the cerebellum, and 4 children had abnormalities on spinal imaging. Although the outcome for most of the children was good, 2 relapsed after cessation of steroids and 5 children had ongoing disabilities. The authors conclude with two points: 1) the investigation-of-choice for establishing the diagnosis of ADEM is MRI of the brain (other investigations were seldom helpful in reaching the diagnosis); and 2) early diagnosis and prompt treatment will probably reduce ADEM morbidity.
Commentary
This study demonstrated that ADEM can be difficult to diagnose secondary to varied clinical presentations and delays in therapy may lead to disabilities. In other studies, the percentage of patients who had residual deficits or long-term disabilities were relatively low; whereas, by the end of this study, 5/18 children continued to have some disabilities. These were not standardized, as other studies have done, with the disability scoring system. Furthermore, the follow-up of patients was not reported in this study so the absolute morbidity could not be summarized. Also, some investigations were not performed in all children because of the retrospective nature of this article. This leads to making broad comparisons difficult and some of the data questionable. However, the validation of MRI scans as a modality to aid in the diagnosis supports previous studies.
MRI to Diagnose, Steroids to Treat
Source: Singhi PD, Ray M, Singhi S, et al. Acute disseminated encephalomyelitis in North Indian children: clinical profile and follow-up. J Child Neurol 2006;21:851-857.
This study evaluated the clinical and neuroradiologic features of ADEM in 52 consecutive patients. The mean age at presentation was 6.14 ± 3.17 years, 73.1% were male, and 17 children had a history of antecedent infectious illness or vaccination. Most children had a meningoencephalitic presentation, with sudden onset motor weakness in 76.9% and seizures in 36.5%. Altered sensorium and pyramidal signs were seen in 55.8% and 80.7% of children, respectively. On MRI, scattered T2-weighted hyperintense lesions were seen, mainly in the subcortical white matter, especially in the parietal (53.8%) and frontal (30.17%) regions. Thalamic, basal ganglia, and callosal lesions were seen in 30.76%, 17.3%, and 13.46% of cases, respectively. Not all 52 children were treated with steroids; 8 children who presented late and had already started showing improvement did not receive specific therapy. Steroid therapy was received by 44 children; 42 received methylprednisolone in a dose of 20-30 mg/kg/day for 5 days. Two patients received dexamethasone (dose was not given). Dramatic recovery after therapy occurred in 26.9% of cases, and marked improvement occurred in 51.9%. In three cases, there was no significant improvement, and in one case, there was deterioration despite therapy. On follow-up, of 44 children, residual smaller MRI lesions were seen in 30. The MRI was repeated in 6 months in children with residual lesions, and it was found that the lesions either disappeared or were significantly reduced after 6 months in 75% of cases. Four children had relapse of ADEM with new lesions on MRI. All of them responded to methylprednisolone. None of the clinical or neuroradiologic factors at presentation had any significant correlation with relapse. Six months after discharge, no deficits could be found in 61.3% of cases; 15.9% and 4.5% had motor and cognitive deficits, respectively; and 9% had multiple deficits.
Commentary
This study demonstrates similar presentation of ADEM in developing countries. The diagnosis can be reliably made with clinical features and MRI. This study also supports the use of corticosteroids; they demonstrated dramatic improvement in a large number of children with ADEM.
Distinguishing Features of MS
Source: Weng WC, Yang CC, Yu TW, et al. Multiple sclerosis with childhood onset: report of 21 cases in Taiwan. Pediatr Neurol 2006;35:327-334.
The records of 21 patients with multiple sclerosis (MS) with onset of symptoms at younger than age 18 were analyzed by investigators at two Taiwanese hospitals. Fifteen patients were female and six male, with a mean age of 12.4 ± 4.5 years at presentation. Presenting symptoms or signs, in order of frequency, were: limb weakness (62%); visual loss or field defect (43%); bulbar symptoms such as dysphagia or dysarthria (33%); sensory disturbance (29%); headache (29%); ataxia (19%); bowel or sphincter dysfunction (14%); and encephalopathy or encephalitis (14%). Multiple symptoms occurred at onset in 76% of patients. A viral prodrome, usually upper respiratory, was reported two weeks before onset of MS symptoms in 43%.
MRIs obtained on all of the patients at the onset of their illness showed lesions in the cerebral white matter in 72%. Lesions were found in periventricular white matter in 56%, and also in basal ganglia (33%), cerebellum (28%), spinal cord (28%), corpus callosum (22%), and optic nerve (17%). Visual evoked potentials were abnormal in 77% of patients, and 62% had optic nerve involvement. Only one patient had optico-spinal MS.
Of 9 patients receiving periodic, subcutaneous interferon beta-1a, four (44%) had no relapses. The course was relapsing/remitting in 86% and progressive in 14%. The mean interval between the first and second attack was 7.2 ± 10 months, most occurring within 12 months of each other. Three patients who were initially diagnosed with ADEM developed MS after 4-month, 2-year, and 6-year intervals, respectively.
Commentary
In the classic reports of MS in U.S. children published in Pediatrics in the 1950s, the symptoms, in order of decreasing frequency, were ataxia or limb weakness, visual disturbance, numbness, headache and vomiting, and urinary incontinence.14 The symptoms were similar to those listed in the current report of MS in Taiwanese children. Although the etiology of MS is unknown, a viral infection early in life, subject to periodic activation, is a hypothesis that is supported by the occurrence of oligoclonal bands in the CSF of MS patients and a viral prodrome reported two weeks before the onset of symptoms in 43% of the patients in this study. The female to male sex ratio of 2.5:1 in the Taiwanese series is similar to that found in most geographic areas and has increased over the past 50 years.14-16
Conclusion/Recommendations
ADEM is usually a single, acute episode of demyelination that can simultaneously affect multiple areas of the CNS. A variety of infections and vaccinations have been associated with ADEM.
Clinical features of ADEM show some consistency. Neurologic deficits attributable to the brain and brainstem develop acutely or subacutely over hours or days, and often in association with more constitutional signs of inflammation and increased intracranial pressure such as fever, lethargy, headache, nausea, vomiting, and meningismus. Brainstem and cerebellar symptoms are common, including ataxia, vertigo, diplopia, dysphagia, and encephalopathy, which can range from obtundation to agitation. Clinical optic neuritis, usually bilateral, or transverse myelitis, usually complete, may be present in association with other impairments.17 Fulminant ADEM, more likely to affect children younger than age 2-3, can progress quickly to coma and impending central or uncal herniation from malignant cerebral edema.2
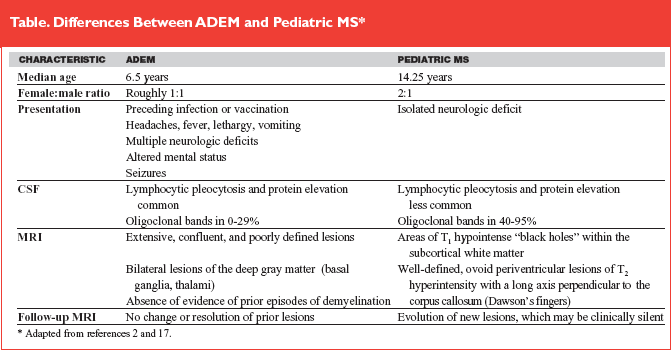
MRI of the brain and spinal cord is the study of choice for the evaluation of patients with suspected demyelinating disease. There are no findings specific to ADEM, but several patterns are recognized, including extensive, confluent subcortical white matter involvement and bilateral deep gray matter involvement. In some instances, the initial MRI is normal, even when neurologic deficits are maximal.18 There should neither be radiologic evidence of past episodes of demyelination on the initial MRI nor evolution of new lesions on follow-up MRI done at least six months later, two findings that are commonly seen in patients with MS. Well-defined, ovoid periventricular lesions whose long axis is perpendicular to the corpus callosum also are more commonly seen in MS. The Table provided reviews the clinical and radiologic distinctions between ADEM and pediatric MS.
 |
Consideration of a possible intracranial infection should lead to a lumbar puncture for CSF analysis. ADEM often will be associated with a mild-to-moderate lymphocytic pleocytosis with a corresponding elevation in CSF protein. The presence of intrathecal IgG synthesis and of oligoclonal bands does not exclude the diagnosis of ADEM, although they are more commonly seen in MS, especially after multiple relapses.
Immunosuppressive or immuno-modulatory therapies are typically used on an empirical basis, without the benefit of prospective research data.17 High-dose IV methylprednisolone is the most commonly given therapy, with most retrospective studies seeing faster recovery and reduced complications in comparison to untreated historical controls.4,17 Theoretically, very high-dose corticosteroids (30-50 mg/kg methylprednisolone) administered intravenously upon presentation to patients with ADEM with or without transverse myelitis, may be advantageous from the vantage point of its capacity to close the blood-brain barrier and limit cerebral or spinal cord swelling.19 This may be particularly true in young children (< 5 years old) who may have marked, permanent neurologic impairments after ADEM.19,20 There are multiple case reports of benefit from IVIG, plasmapheresis, or cytotoxic agents in patients who did not initially respond well to other therapies.4,17
Recent case series report excellent recovery in up to 90% of children with ADEM, and there is surprisingly little predictive value to the severity of the neurologic deficits at presentation or nadir. Many of the children with a near-complete recovery have only mild visuospatial processing deficits that manifest as difficulty in school.
Some patients will undergo an early relapse of their initial symptoms, with flare-up of the previously involved areas of the CNS on repeat neuroimaging within four weeks of initial presentation. This can occur as steroid therapy is tapered or stopped. In some series with long-term follow-up, up to 35% of patients undergo one or more distinct recurrences, with new neurologic deficits and areas of demyelination developing more than one month after the resolution of a prior episode. Late follow-up neuroimaging to assess for the development of new asymptomatic lesions 3-6 months after initial diagnosis may be particularly useful in the identification of patients who are at the highest risk for progression and relapse. There is ongoing debate as to whether ADEM and MS are distinct disorders or coexist on a spectrum of autoimmune demyelination. Despite the advances in our understanding of pathogenesis of inflammatory demyelinating CNS disorders, the only truly reliable diagnostic test remains time.3
References
1. Silvia MT, Licht DJ. Pediatric central nervous system infections and inflammatory white matter disease. Pediatr Clin North Am 2005;52:1107-1126.
2. Michelson DJ. Acute disseminated encephalomyelitis. In: Perkin RM, Swift J, Newton D, Anas N, eds. Pediatric Hospital Medicine. Philadelphia: Lippincott Williams and Wilkins; 2007:296-298.
3. Dale RC, Branson JA. Acute disseminated encephalomyelitis or multiple sclerosis: can the initial presentation help in establishing a correct diagnosis? Arch Dis Child 2005;90:636-639.
4. Tenembaum S, et al. Acute disseminated encephalomyelitis. Neurology 2007;68(Suppl 2):S23-S36.
5. Stonehouse M, et al. Acute disseminated encephalomyelitis: recognition in the hands of general paedatricians. Arch Dis Child 2003;88:122-124.
6. Bennetto L, Scolding N. Inflammatory/post-infectious encephalomyelitis. J Neurol Neurosurg Psychiatry 2004;75(Suppl I):i22-i28.
7. Ness JM, et al. Clinical features of children and adolescents with multiple sclerosis. Neurology 2007;68(Suppl 2):S37-S45.
8. Gadoth N. Multiple sclerosis in children. Brain Dev 2003;25:229-232.
9. Ozakbas S, et al. Childhood and juvenile onset multiple sclerosis: Clinical and paraclinical features. Brain Dev 2003;25:233-236.
10. Dale RC, et al. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 2000;123(pt 12):2407-2422.
11. Confavreux C, et al. EDMUS, a European database for multiple sclerosis. J Neurol Neurosurg Psychiatry 1992;55:671-676.
12. Krupp LB, et al. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007;68(Suppl 2):S7-S12.
13. Honkaniemi J, et al. Delayed MR imaging changes in acute disseminated encephalomyelitis. AJNR Am J Neuroradiol 2001;22:1117-1124.
14. Millichap JG. Clinical manifestations of childhood multiple sclerosis. AAP Grand Rounds 2007;17:30-31.
15. Renoux C, et al. Natural history of multiple sclerosis with childhood onset. N Eng J Med 2007;356:2603-2613.
16. Mikaeloff Y, et al. Prognostic factors for early severity in a childhood multiple sclerosis cohort. Pediatrics 2006;118:1133-1139.
17. Menge T, et al. Acute disseminated encephalomyelitis: an update. Arch Neurol 2005;62:1673-1680.
18. Khurana DS, et al. Acute disseminated encephalomyelitis in children: discordant neurologic and neuroimaging abnormalities and response to plasmapheresis. Pediatrics 2005;116:431-436.
19. Rust RS. Acute disseminated encephalomyelitis. Emedicine http://www.emedicine.com/neuro/topic500.htm. Accessed 10/8/07.
20. Jacobs RK, et al. Neuropsychological outcome after acute disseminated encephalomyelitis: impact of age at illness onset. Pediatr Neurol 2004;31:191-197.
Defined as an inflammatory demyelinating disease process that affects the brain and spinal cord, acute disseminated encephalomyelitis (ADEM) has been called the most common white matter disease in children.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.