Stroke in Young People: A Different Differential
Stroke in Young People: A Different Differential
Authors: Susan P. Torrey, MD, FACEP, Associate Residency Director, Department of Emergency Medicine, Baystate Medical Center, Springfield, MA; and Tala Elia, MD, Attending Physician, Department of Emergency Medicine, Baystate Medical Center, Tufts University School of Medicine, Springfield, MA.
Peer Reviewers: Jonathan Glauser, MD, FACEP, Chairman, Emergency Department, Cleveland Clinic, Cleveland, OH; and Steven M. Winograd, MD, FACEP, Attending Physician, Emergency Department, Horton Hospital and Arden Hill Hospital, Orange County, NY.
I trained during the era when the prevailing approach for patients with acute strokes was to define the area of brain affected using only clinical examination, exclude treatable intracranial hematoma (without CT!), and to admit the patient for a period of observation and initiation of physical rehabilitation. With the exception of possibly using heparin for crescendo TIAs, there was a nihilistic attitude about stroke treatment. Now we have prehospital stroke triage criteria, primary stroke centers, acute stroke teams, and more imaging technologies than I can remember. In addition, we are discovering more causes for acute strokes, especially in younger patients. This manuscript reviews these causes, their assessment, and management. After reading this, I suspect many will revise their acute stroke protocols and order sets to consider these possibilities in younger patients.
J. Stephan Stapczynski, MD, FACEP, Editor
While stroke is most often a result of progressive atherosclerotic cerebrovascular disease, occurring with increasing frequency as the population ages, there are less common causes of stroke that mainly affect younger populations. These alternative etiologies of stroke symptoms require an emergency physician to have a broader differential diagnosis of acute focal neurologic symptoms in a young population, and sufficient knowledge to facilitate an appropriate evaluation and treatment plan. The differential diagnosis of stroke in young people (usually defined as younger than 45 years) includes dissection of cerebrovascular arteries; thrombosis of cerebral veins and venous sinuses; embolism in general, and paradoxical embolization via a patent foramen ovale in particular; some causes of intracerebral hemorrhage; and a list of unique causes.
Epidemiology
Epidemiologic studies of strokes in young adults have been limited by population studies that use highly variable diagnostic criteria and evaluation protocols. Given these limitations, it remains common to find many of these patients with no specific etiology for their ischemic stroke, hence the term "cryptogenic stroke" which has come into popular use in the last decade. A review of population-based articles1-5 finds that strokes in young people (under the age of 45) have an incidence of 10-34 cases per 100,000 population per year. Of these, the most common etiologies include premature atherosclerotic vascular disease (7-11%), hematologic disorders inducing thrombophilia (7%), cervical artery dissection (8-20%), cardioembolic (6-33%), and cryptogenic stroke (21-60%). Unlike older patients diagnosed with ischemic stroke, younger patients generally have little to no risk factors for atherosclerotic disease. Diagnostic testing for vascular disease tends to be unremarkable and does not reveal the cause of the cerebral infarct in the vast majority of young adult patients. Up to 43% of young adults with ischemic strokes have no identifiable cause and are classified as "cryptogenic strokes." Not surprisingly, the study with the lowest number of cryptogenic strokes had the most rigorous diagnostic plan, including liberal use of CT and MRI with angiographic evaluation of carotid arteries and posterior circulation, as well as frequent transesophageal echocardiography.1
 |
Patent Foramen Ovale
The high rates of cryptogenic stroke in young, seemingly healthy adults have led to investigations for other causes of stroke beyond vascular and atherosclerotic disease. One strong association that has emerged is the relationship between cryptogenic strokes and a patent foramen ovale (PFO). A PFO is a remnant of fetal circulation that results from the failure of the primum and secundum septa to fuse. The prevalence of PFOs in the general population is as high as 27%, affecting men and women equally.6 Patients younger than 55 years old with a cryptogenic stroke have an associate with PFO six times greater than a control population, leading to the hypothesis that a PFO may play a role in the cause of stroke in these patients.7
The proposed mechanism for the involvement of PFOs in stroke is by migration of a thrombus from the venous circulation to the right atrium. The clot is then shunted to the left atrium via the PFO and from there embolizes systemically.8 This phenomenon is also known as a "paradoxical emboli" and occurs when right atrial pressure exceeds left atrial pressure. In a patient with normal cardiovascular function, this right to left shunt can be transient during normal respiration or after the release of Valsalva maneuvers such as straining or coughing. Fourteen percent of asymptomatic patients with a PFO have a right to left shunt at rest. That number increases to 23% with Valsalva maneuvers.9 Despite this, patients with cryptogenic stroke and PFO rarely report symptom onset after valsalva maneuvers. Other causes of a right to left shunt include acute pulmonary embolism, which causes pulmonary hypertension, as well as right ventricular infarct and acute respiratory failure.8 One study even reports an incidence of paradoxical embolism as high as 60% in patients with pulmonary embolism.10 Therefore, when confronted with an ischemic stroke in a young adult in which a paradoxical embolism is suspected, consideration should be given to determine if an identifiable embolic source or precipitating factors exist. However, in most patients the source of thrombus remains unidentified despite diagnostic testing.
Several factors are thought to increase the risk of stroke in patients with a PFO. These include a large opening, a large right to left shunt or a right to left shunt at rest, a diagnosis of pulmonary embolism, and the presence of an atrial septal aneurysm (ASA).8 In fact, presence of both a PFO and ASA showed a 33-fold increase in cryptogenic stroke over the general population.11 Patients with an ASA and PFO concurrently tend to have large PFOs, which may contribute to this increased risk.12-14
The diagnostic test of choice in patients with suspected PFO is transesophageal echocardiography (TEE). In one study, TEE was able to find the source of cardioembolism in 57% of patients with cryptogenic stroke as opposed to only 10-15% with transthoracic echocardiography.15 Injection of agitated saline contrast into a peripheral vein can aid in the detection of a PFO via TEE by quantifying the number of bubbles that migrate from the right to the left atrium after three cardiac cycles.16 (See Figure 1.)
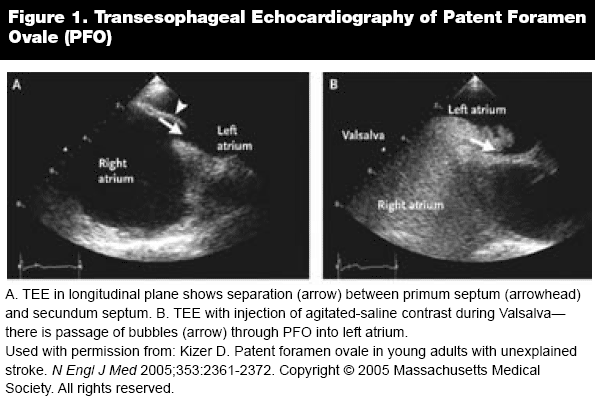 |
The risk of recurrence of stroke in young patients who have a PFO is proportional to the size of the opening, the degree of shunting, and the presence of an ASA. However, the general rate of recurrence tends to be low in patients younger than the age of 60, ranging from 1.9% to 2.3%.17,18
Patients with a patent foramen ovale who have had a stroke have several therapeutic options, but the data comparing these options is limited. One treatment is anticoagulation with an antiplatelet agent, such as aspirin, clopidogrel, or dipyridomole. An alternative is treatment with warfarin. The data differentiating between aspirin, as opposed to warfarin, for anticoagulation is inconclusive but there was a slight increased risk of bleeding with warfarin.18 Other options include direct surgical closure and percutaneous closure. It is unclear whether either of these closure techniques is superior to anticoagulation, but percutaneous closure is only FDA-approved for cases of recurrent stroke despite therapeutic oral anticoagulation.19
Cerebrovascular Dissection
The annual incidence of spontaneous carotid artery dissection is 2.5-3 per 100,000, and the estimated frequency of vertebral artery dissection is 1-1.5 per 100,000. While dissection of the vertebral or carotid arteries accounts for only 2% of all ischemic strokes, they are an important cause of stroke in young people, and account for 10-25% of such cases.20 Dissections of the cervical arteries affect all age groups, including the pediatric population, but there is a definite peak in the fifth decade. Men and women are affected equally, although women are on average five years younger at the time of dissection.
The pathophysiology of arterial dissection involves an intimal tear in the vessel, which allows blood to enter into layers of the arterial wall and form an intramural hematoma.20 Such defects may follow significant trauma or occur spontaneously, although most dissections probably involve some minor trauma or mechanical stress to the artery.21,22 Extracranial segments of the carotid and vertebral arteries experience dissection more often than intracranial segments or similar sized arteries elsewhere in the body. This increased prevalence in the arteries of the neck may be explained by the greater mobility of these extracranial segments and increased exposure to trauma. Once blood enters the arterial wall, the hematoma may extend either toward the intima or the adventitia. Subintimal dissection tends to cause stenosis of the vessel lumen, while subadventitial dissection may cause aneurismal dilation of the artery. The expanding intramural hematoma may cause arterial lumen compromise with hypoperfusion and ischemia, however the most common etiology of cerebral ischemia associated with dissection is the result of embolism of thrombotic material adherent to the narrowed lumen at the site of dissection.23,24
The pathogenesis of cerebral artery dissection is likely an interaction of genetic and environmental factors.21 Most people experiencing an arterial dissection have an underlying structural defect of the artery wall, although the exact arteriopathy is impossible to define in most cases. Well described and inherited disorders associated with arterial dissection include Ehlers-Danlos syndrome, Marfan's syndrome, autosomal dominant polycystic kidney disease, as well as fibromuscular dysplasia and cystic medial necrosis. Genetic studies are on-going to further define inherited risk.25
Given a genetic predisposition, an inciting trauma or stress to the artery may cause cervical artery dissection. Direct blows to the neck, hyperextension, and even spinal manipulative therapy have been described as independent risk factors for dissection.26,27 Spinal manipulative therapy may simply be coincidental, as patients seek manipulative therapy for neck and head pain, which may be the initial symptom of dissection. Certainly, it is prudent to consider cervical dissection in any patient with new or increased neck pain or the development of neurologic signs within days of manipulative therapy.26 Finally, a recent case report raises the concern about arterial dissection and stroke after methamphetamine and cocaine use.28
The typical patient with a carotid artery dissection presents with pain on one side of the head, face, or neck accompanied by a partial Horner syndrome (oculosympathetic palsy) and followed by cerebral or retinal ischemia with hours or days. This classic triad of symptoms is found in fewer than one-third of patients,20 but the presence of two of the three symptoms should strongly suggest the diagnosis. Pain is the most frequent manifestation of carotid dissection, occurring in 80%,29 and is most often described as severe, constant, and throbbing. In a series where imaging of the cervical arteries was liberally applied, pain was the only symptom of cervical artery dissection in 8% of patients.30 Oculosympathetic palsy, or a partial Horner syndrome, includes ipsilateral miosis and ptosis as sympathetic nerve fibers along the internal carotid artery are disrupted. Anhidrosis, the third component of a classic Horner syndrome, is not seen with dissection of the internal carotid, as facial sweating is innervated by sympathetic fibers from the external carotid artery. Other symptoms of the dissection itself may include pulsatile tinnitus (25%), a bruit audible to the patient because of proximity of the carotid to the ear and cranial nerve palsies (12%), especially of the hypoglossal nerve.21 Intraluminal thrombotic material may subsequently embolize, causing transient ischemic attacks (TIAs) or cerebral infarcts (CVA), with symptoms including amaurosis fugax, hemiplegia, or dysphasia. Dissection of the internal carotid almost always causes deficits in the MCA distribution.24
Vertebral artery dissection typically presents with pain in the posterior neck or occipital head followed by ischemia in the posterior circulation. Neurologic symptoms may include pain or weakness in an arm as a result of cervical root involvement or brain stem infarcts, particularly of the lateral medulla leading to a Wallenberg's syndrome (including dysphagia, diplopia, Horner syndrome, vertigo, nausea, and vomiting).31
Diagnosis of cervical artery dissections requires first and foremost a high index of suspicion. Catheter angiography was the gold standard diagnostic test for many years. The most common finding on angiography is the so-called "string sign" a long segment of narrowed lumen. Currently, MRA has essentially supplanted this invasive technique.32 Intramural hematomas are shown as hyperdense signals on T1 weighted imaging and characteristically have a crescent shape adjacent to the lumen. (See Figure 2.) The need to rule out arterial dissection in the cervicocranial arteries is on a short list of indications for emergent MRI.33 Increasingly, helical CT angiography is being recognized as an adequate diagnostic test, and thus the choice of diagnostic test is best made after discussion with local neurology and radiology to define the most appropriate, available test.
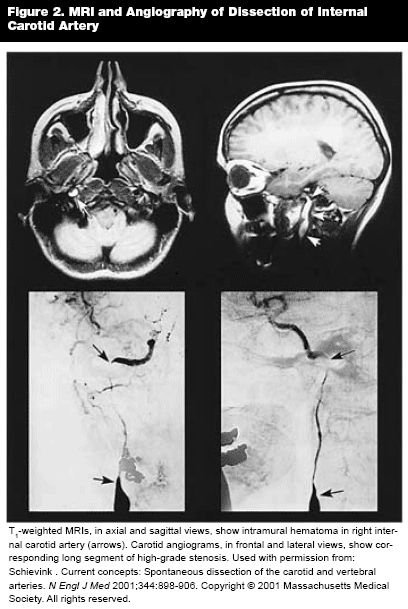 |
Because the cerebral ischemia associated with dissection of the carotid or vertebral artery is caused by embolization in the vast majority of cases,21 antiplatelet therapy or anticoagulation is, in fact, the treatment of choice. Standard treatment of arterial dissection includes heparinization until adequate anticoagulation with warfarin is accomplished, and maintaining an INR of 2.5-3.0 for 3-6 months.34 Contraindications to anticoagulation include the presence of a large infarct with mass effect, hemorrhagic transformation of the infarct, and intracranial extension of the dissection, in which case antiplatelet therapy with aspirin (325 mg a day) is recommended. Anticoagulation or antiplatelet therapy is continued until a followup MRA at 3 or 6 months reveals healing of the intraluminal abnormalities.34
It is also possible to safely treat strokes secondary to arterial dissection with thrombolytics.35,36 Local complications from extension of the intramural hematoma do not occur. Given current urgency to administer thrombolytic to acute stroke syndromes, particularly in younger patients with appropriate presentations and a negative head CT, these reports of the safety of thrombolysis in dissection are reassuring.
Generally, the prognosis of cervicocerebral artery dissection is quite good. The death rate from dissection of the carotid and vertebral arteries is less than 5%. Almost 70% of patients make a good recovery with no or little neurologic deficit. The risk of recurrent dissection in an unaffected artery is about 2% during the first month but then decreases to about 1% per year.20
Cerebral Venous Thrombosis
The spectrum of presentations for cerebral venous thrombosis (CVT) is diverse and can range from an isolated headache to altered mental status with focal neurological deficits. The wide, and sometimes subtle, range of presentations of CVT can lead not only to a missed diagnosis but also to a failure to diagnose a venous thrombus as the underlying cause of what appears to be a stroke in a young adult. Therefore, it is important as emergency medicine practitioners to understand the possible presentations of cerebral venous thrombosis as well as the causes and risk factors associated with this diagnosis.
Cerebral venous thrombosis is an entity that tends to affect young and middle-aged adults, with some predominance toward females. The underlying cause of CVTs can sometimes be traced to an underlying coagulopathy. Hypercoagulability factors such as factor V Leiden, antithrombin III deficiency, and protein C or protein S deficiency have all been known to play a role in the development of venous thrombosis.37
Pregnancy has long been described as an inciting factor for cerebral venous thrombosis with an increased incidence during the peripartum and postpartum period. The highest incidence seems to be in the postpartum period, and risk factors include hypertension in pregnancy and cesarean delivery.38,39 The prognosis of peripartum CVT, however, tends to be better than that of CVT associated with other causes.40
The use of oral contraceptive pills (OCP) increases the risk of both venous and arterial thrombus in certain patients, and an increased risk of cerebral venous thrombosis is no exception.41,42 The use of OCPs as the precipitant for cerebral venous thrombosis is more common in developed countries and also in women who have a concomitant coagulopathy or history of thrombosis.
Septic thromboses account for up to 10% of cerebral venous thrombosis and are a common cause of cavernous sinus thrombosis.42 They are a well described complication of bacterial sinusitis and other face or neck infections. As opposed to inflammatory and coagulative causes, the onset of septic thrombosis tends to be more acute and carry a worse prognosis.38
Many factors affect the clinical presentation of cerebral venous thrombosis including patient age, the location and extent of the thrombus, and the rate of propagation of the thrombus. Headache is present in 80-95% of patients.38 The headache tends to be severe, progressive, and persistent, but there is no classic manifestation of the headache and it can also present suddenly with either diffuse or localized pain.43 About half of the patients with a diagnosis of CVT have an altered mental status, while 15% are comatose. Seizures, both partial and generalized, are common and found in more than 40% of patients. Focal neurological deficits, both motor and sensory, occur in 30-40% of patients. The unusual finding of focal deficits alternating from one side to another actually can be a late indication of a superior sagittal sinus thrombus. Another common finding is papilledema secondary to increased intracranial pressure, found in up to 50%.44 (See Table 2.)
 |
Depending on the site and acuity of the thrombus, these many symptoms generally coalesce into one of four clinical patterns.38 Focal neurological deficits and/or parietal seizure predominate in the first of these patterns. This pattern is the one that is most likely to mimic the presentation of an acute ischemic stroke. In addition to motor or sensory deficits in the extremities, these patients may also have a headache and altered mental status. One exception is the rare incidence in which the cortical veins are involved but thrombus does not extend into the dural sinus. These patients have no signs of increased intracranial pressure, and their symptoms are very similar to an ischemic stroke. The second presentation, that of isolated increased intracranial pressure, can be mistaken for idiopathic intracranial hypertension and presents with headache, nausea, vomiting, and papilledema. This presentation may be more chronic, and late signs may include transient visual losses and eventually sixth nerve palsy.38 A venous thrombus may also present itself as subacute diffuse encephalopathy. These patients tend to have no definitive localizing signs but have a decreased level of consciousness and occasionally seizures. They present similarly to a patient with encephalitis or a systemic metabolic disturbance. The last pattern, painful ophthalmoplegia or cavernous sinus syndrome, is more distinct. This presentation is a constellation of symptoms that usually results from a septic thrombus. The thrombus originates either from direct extension from sinusitis or from hematogenous spread of a face or neck infection. These patients have proptosis, chemosis, painful extraocular movements, papilledema, and deficits of the third, fourth, or sixth cranial nerves.41
Although computed tomography (CT) is the most readily available and utilized imaging modality for acute intracranial processes in the emergency department, it is unfortunately not the study of choice for diagnosis cerebral venous thrombosis. Some patients may have positive CT findings such as the "empty delta sign" in which a superior sagittal sinus clot is outlined by contrast filling of collateral veins or the "dense triangle sign" in which a new clot in the superior sagittal sinus is actually visualized on a noncontrast CT. However, despite the presence of these signs and other non-specific findings of hypodensities and blood from a secondary hemorrhage, 30% of patients will have a normal head CT.38,45
Magnetic resonance imaging (MRI) is the study of choice for the diagnosis of CVT.38,42,45 MRI has the benefit of being able to visualize the clot directly but can be limited in the very early stages of the thrombus. In these cases, magnetic resonance venography (MRV) is particularly useful, with accuracy for diagnosing venous thrombosis approaching 100%.43 (See Figure 3.) It has replaced conventional angiography in many institutions because of its availability and noninvasiveness. Helical CT venography is also becoming more readily available. CT venography has less artifact and generally can be obtained more quickly than MRV. In comparison to MRV, CT venography has been shown to have an accuracy of 90-100%.46-48
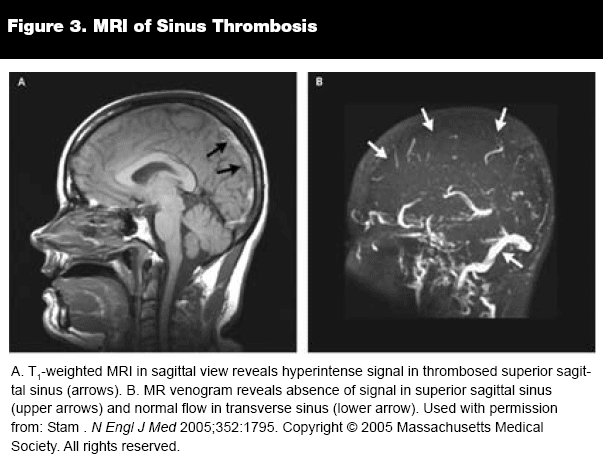 |
In some cases a lumbar puncture may be performed in these patients to rule out other potentially dangerous causes of headache such as subarachnoid hemorrhage and meningitis. While cerebrospinal fluid analysis is non-diagnostic for CVT, 50% have increased protein, 60% have increased RBCs, and 30% have leukocytosis.44
Heparin is the treatment of choice in patients diagnosed with cerebral venous thrombosis. It has been studied extensively and has been shown to be a safe and effective treatment, even in those patients with a secondary hemorrhagic infarct.45,49,50 As with other types of venous thrombosis, heparin therapy eventually can be switched to oral warfarin. If the patient has no underlying coagulopathy, warfarin treatment should continue for 6 months to maintain an INR between 2.0-3.0.45
There has been discussion in the literature on the use of endovascular thrombolytics in these patients. However, although venous flow may be restored more quickly with the use of thrombolytics, there has been no evidence that there is any improvement in clinical outcome. There is an increased risk of hemorrhagic complications associated with thrombolytics when compared to heparin.45 Considering the limited data regarding thrombolytics in this setting, their use should be restricted, but may be indicated in those cases in which there is decompensation despite therapeutic anticoagulation.
Several treatment options exist for those patients who have increased intracranial pressure (ICP) associated with CVT and they do not differ drastically from the treatment of increased ICP from other causes. In some cases, treatment of the thrombus with heparin alone will aid in the lowering of the intracranial pressure. Other patients with severe intracranial hypertension may require additional treatment with acetazolamide, mannitol, or glycerol. The role of corticosteroids in this setting is controversial and data is limited regarding their benefit.38 Patients with vision-threatening papilledema may need one or more lumbar punctures to drain cerebrospinal fluid and decrease intracranial pressure.51 In patients with refractory papilledema, optic nerve fenestration may be indicated. Ventriculoperitoneal shunts and, in rare cases, decompressive craniotomy may be necessary for severely increased intracranial pressure.
Although seizures are not uncommon, the use of seizure medications is not indicated prophylactically for all patients. Anti-convulsants should be started in patents who have already had a seizure or in those with a cerebral lesion on CT or MRI.45
Coagulopathies
In young patients who present with an ischemic stroke or cerebral venous thrombus and no clear identifiable cause, an investigation into underlying coagulopathies is sometimes warranted. The yield of coagulopathic testing for all stroke patients is low; however the yield significantly increases when patients younger than the age of 50 years are selected for testing.52,53
Because the coagulopathies associated with venous thrombosis differ from those associated with arterial thrombosis, it is important to distinguish between patients with a venous thrombus as a source of their deficit, such as cerebral venous thrombosis or a paradoxical embolism, versus those presenting with an ischemic stroke originating from an arterial thromboembolism.54 In patients with venous thromboembolism, factor V Leiden, as well as protein C deficiency, are the most commonly encountered underlying coagulopathies.54 This differs from patients diagnosed with an ischemic stroke secondary to an arterial thrombus. The commonly diagnosed coagulopathies associated with ischemic strokes are antiphospholipid antibodies (also referred to as lupus anticoagulant or anticardiolipin antibodies), heparin antibodies, and elevated homocysteine level.52,55 Other coagulopathies that patients may be screened for include antithrombin III deficiency, protein S deficiency, plasminogen deficiency, and prothrombin gene mutations.54
It is important that patients who initially screen positive for a coagulopathy such as protein C or S deficiency, antithrombin III deficiency, or anticardiolipin antibodies receive appropriate follow-up. Not only do these patients require longer-term anticoagulation but they should also be re-tested 2-3 months after the thrombotic event as initial laboratory tests may be falsely abnormal during the acute phase of a thrombus.55
Oral Contraceptive Pills
With more than 10 million women in the United States and 78.5 million women worldwide using oral contraceptive pills (OCPs), they are an important stroke risk factor.56 The use of OCPs not only increases the risk of venous thromboembolism, but also the risk of ischemic stroke. Traditional oral contraceptive pills are associated with nearly a 3 times increased risk.56 Although this risk is decreased with the use of newer OCPs that contain one-third to one-fifth the amount of ethinyl estradiol, some studies argue an increased risk still exists.56,57 Co-factors that further compound the risk of stroke or thromboembolism includes hypertension, smoking, age over 35, and migraine headaches.56,57
Pregnancy
Stroke is a long described and potentially devastating complication of pregnancy. It can manifest as cerebral infarction, either venous or arterial, or as an intracranial hemorrhage.58,59 The incidence of stroke during pregnancy still remains relatively rare, affecting about 24 of every 100,000 deliveries and the mortality rate, 4.1-14%, is lower than that of strokes in general.60 Nonetheless, these events can still be debilitating. In one retrospective analysis, 14-22% of these patients were disabled enough to be discharged to another facility rather than home.60 Factors that increased the risk of stroke during pregnancy or postpartum include hypertension, preeclampsia, concurrent infection, cesarean section, African-American race, and age greater than 35.58,60 The time period in which the risk of pregnancy-related stroke is the greatest is not actually during pregnancy but during the six weeks postpartum.61
Spontaneous Intracerebral Hemorrhage
Intracerebral hemorrhage (ICH) accounts for 10-15% of all cases of stroke, and is associated with the highest mortality rate, with only 38% of patients surviving one year.62 While ICH is not more prevalent in a general younger population, there are several considerations that recommend that this diagnosis be included in a discussion of stroke in young people, including risk factor assessment and the extent of diagnostic evaluation.
Primary intracerebral hemorrhage, which accounts for more than 80% of cases, occurs as a result of spontaneous rupture of small vessels damaged by chronic hypertension. Hypertension increases the risk of intracerebral hemorrhage, particularly in patients who are not compliant with medications, are 55 years of age or younger, or are smokers.63,64 Improved control of blood pressure improves the incidence of hemorrhage, as does smoking cessation. Modifications of these risk factors are especially relevant in several genetic populations, including blacks and Japanese, who have a significantly increased risk of intracerebral hemorrhage (50-55 per 100,000 population, compared with 10-20 per 100,000 for overall worldwide incidence). Other important risk factors for intracerebral hemorrhage include alcohol and drug abuse, which are also pertinent to a younger population. Amphetamine abuse is associated with an increased risk of intracerebral hemorrhage, while cocaine use appears to increase the incidence of both hemorrhagic and ischemic strokes.65
Phenylpropanolamine, a synthetic sympathomimetic amine, gained notoriety in 2000 following a study that suggested that its inclusion in appetite suppressants, and possibly in cough and cold remedies, was an independent risk factor for hemorrhagic stroke in young women.66 To put the risk into perspective, this article estimates that one woman may have a stroke due to this drug for every 107,000-3,268,000 women who use products with phenylpropanolamine as an appetite suppressant within a three-day window. In light of this study, phenylpropanolamine-containing products were removed from drugstore shelves.
There is an increasing incidence of anticoagulant-associated intracerebral hemorrhage. A recent study found the incidence has quintupled during the 1990s,67 largely due to increasing use of warfarin for prevention of strokes associated with atrial fibrillation. The current incidence of anticoagulant-associated ICH approaches that of subarachnoid hemorrhage (6.6 cases per 100,000 persons), and accounts for 8-14% of all intracerebral hemorrhages.68 Intracerebral hemorrhage with anticoagulation is also associated with a poor prognosis. The outcome is fatal in two-thirds of these patients with INR greater than 3.0 at presentation.
A final risk factor for intracerebral hemorrhage that is pertinent to a young population is the increased risk during pregnancy and postpartum (6.1 pregnancy-related ICH per 100,000 deliveries).69 Intracerebral hemorrhage accounts for a substantial portion of pregnancy-related mortality. The risk of ICH associated with pregnancy is greatest in the postpartum period, and independent risk factors include advanced maternal age, African American race, hypertensive disease, coagulopathy, and tobacco abuse.
Patients with intracerebral hemorrhage typically present with the abrupt onset of severe headache and developing neurologic deficits, or with decreased level of consciousness. Patients with a supratentorial intracerebral hemorrhage involving putamen, caudate, and thalamus have contralateral sensory-motor deficits due to involvement of the internal capsule. Patients with infratentorial bleeds will have signs of brain stem dysfunction, such as abnormalities of gaze, cranial nerve abnormalities, and contralateral motor deficits. Patients with hemorrhage within the cerebellum develop ataxia, nystagmus, and dysmetria. Nonspecific symptoms typically include headache and vomiting due to increased intracranial pressure and meningismus when blood extends to the ventricles. One-fourth of patients with intracerebral hemorrhage who initially are alert will have deterioration in level of consciousness within the first 24 hours after the onset of bleeding, usually associated with continued or recurrent bleeding. (See Figure 4.)
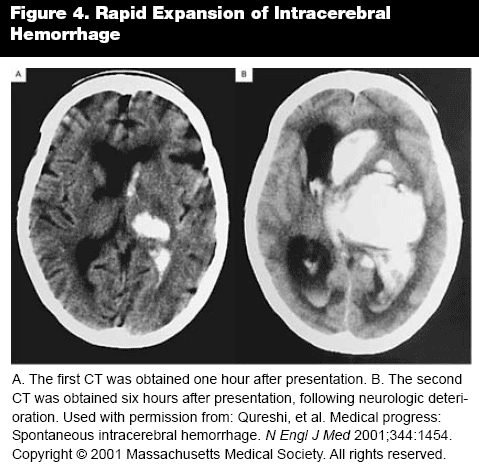 |
Intracerebral hemorrhage is diagnosed easily and reliably by non-contrast CT scan of the head. The location and size of the hematoma, the presence of interventricular blood, and the occurrence of hydrocephalus should be noted. Selected patients may need to undergo angiography (catheter angiography, or contrast enhanced MRI or helical CT) to look for secondary causes of hemorrhage, including aneurysms, arteriovenous malformations, and vasculitis. Nearly half of patients who are normotensive and 45 years of age or younger had abnormalities by angiography, whereas hypertensive patients older than 45 years rarely had a secondary cause of the bleed. The American Heart Association guidelines recommend angiography for all patients with no clear cause of hemorrhage who are candidates for surgery, particularly young patients without hypertension who are clinically stable.70
The medical management of intracerebral hemorrhage in the emergency department initially includes evaluation of the airway and decisions about intubation. This becomes a critical action for patients with decreased level of consciousness or impairment of airway protective reflexes. Approximately 30% of supratentorial hemorrhage, and virtually all patients with a brain stem or cerebellar hemorrhage have altered level of consciousness and will require intubation.62 While aggressive treatment of hypertension in the setting of an acute intracerebral hemorrhage remains controversial, most authors agree that in the hypertensive patient (MAP > 130 mmHg), it is reasonable to decrease MAP by 15% with labetalol or nicardipine infusion. Finally, correction of a coagulopathy is of paramount importance. For the patient who has been using warfarin, rapid correction with fresh frozen plasma (15 mL/kg body weight) and vitamin K (2-10 mg IV) is standard care.71 Unfortunately, vitamin K takes 6-24 hours to normalize the INR. Fresh frozen plasma, in volume sufficient to reverse a severe coagulopathy (often 2-4 liters) can be a limiting factor in seriously ill patients. More recent trends in the treatment of warfarin-associated coagulopathy include the use of prothrombin complex concentrates (PCC – factor IX concentrate) and recombinant factor VIIa (rFVIIa).72
For the patient with a Glasgow Coma Scale (GCS) score of < 8, or with neurologic deterioration, emergent treatment of increased intracranial pressure is appropriate, and urgent neurosurgical consult for placement of an intracranial pressure (ICP) monitor is indicated. Temporizing measures that will decrease ICP until further interventions are available include hyperventilation and the use of mannitol (0.25-1.0 g/kg body weight).71 Hyperventilation to a PCO2 of 25 to 30 mmHg is highly effective and will rapidly lower ICP, but its effects are short-lived. Mannitol will have peak effects in approximately 20-30 minutes and may have a duration of action of several hours.
Surgical treatment of intracerebral hemorrhage remains controversial, although it is possible to state a few truths and review the current standard of care.70-73 While neurosurgical consultation is appropriate for any patient with ICH, the urgency with which it needs to be accomplished will vary. For the patient with hydrocephalus on CT or secondary neurologic deterioration due to intraventricular blood, an external ventricular drain is appropriate. Ventriculostomy also allows ICP monitoring, which will facilitate treatment of increased ICP. For the patient with a cerebellar hemorrhage > 3 cm or hemorrhage of any size with neurologic deterioration, surgical evacuation is indicated. Finally, any large accessible cortical hematoma or secondary neurologic deterioration in a young patient mandates consideration of a neurosurgical procedure.71
The prognosis of intracerebral hemorrhage is poor, with 23-58% mortality at six months. Several factors are predictive of higher mortality a low initial GCS, a large volume hematoma, and the presence of blood in the ventricular system.
Miscellaneous Causes of Stroke in Young People
Other considerations with stroke in young people include associations with HIV infection, cancer, sickle cell disease and trait, illicit drug abuse, and migraines. Finally, this discussion includes conversion reactions, which must be considered in the presentation of many young people with otherwise unexpected neurologic deficits.
Acquired immunodeficiency syndrome (AIDS) is strongly associated with stroke,74 with one study reporting an increased adjusted relative risk of 14% for ischemic stroke and 25% for intracerebral hemorrhage.75 While some have postulated a primary angiitis of the CNS associated with HIV infection,76 it is clear that many strokes in these patients are secondary to associated infectious and neoplastic processes associated with AIDS.
Strokes, including hemorrhage and infarction, are second only to metastases in frequency of CNS lesions in autopsy series of patients with cancer. A recent large retrospective review of cancer patients with cerebral infarction found causes of infarction evenly distributed between embolic and nonembolic,77 and a suggestion that a hypercoagulable state was a leading risk of stroke in this population, as evidenced by nonbacterial thrombotic endocarditis and other cardioembolic sources. Also noted was a higher frequency of stroke in patients with lung and primary intracranial cancer. Once stroke occurred in a patient with cancer, regardless of etiology, the overall prognosis was poor, with median survival of only 4.5 months.77
Stroke is a devastating complication of sickle cell disease and occurs in approximately 11% of those with hemoglobin SS younger than the age of 20, and may be associated, with intracranial hemorrhage, especially in the adult population.78 In response to chronic anemia and hypoxemia, cerebral blood flow is markedly increased in sickle cell disease, as evidenced by increased velocity in major cerebral arteries by transcranial Doppler ultrasonography. When acute conditions arise leading to diminished oxygen availability, the increased hypoxic stress leads to ischemia, typically in the parenchymal areas of the anterior and middle cerebral arteries and the border zones between their distal circulations. It has even been suggested that sickle cell trait is a potential risk factor for stroke in the African-American population.79
The relationship between migraine and stroke has been shown by epidemiologic studies, and most conclude that a history of migraine is an independent risk for ischemic stroke.80,81 A migranous infarct in which the pathogenesis is directly due to migraine is diagnosed when the stroke occurs during a typical migraine with aura and when other causes have been excluded. While this etiology of stroke in young people continues to appear in lists of population studies, it is probably wise to limit the use of this diagnosis, at least until an extensive search for other causes has been exhausted.
Finally, conversion reaction can present with a very convincing constellation of neurologic symptoms, which may appear to be an acute stroke. Typically there is the sudden onset of a single symptom, often simulating some nonpainful neurologic disorder for which there is no pathophysiologic explanation. The neurologic symptoms are not under the patient's voluntary control, the symptoms are often associated with antecedent stress (and occasionally have some symbolic relationship to the precipitant), and they may be associated with secondary gains. Almost one-third of patients with conversion reaction have a history of previous psychiatric diagnosis. La belle indifference refers to an apparent lack of concern by the patient for their symptoms, and has long been regarded as typical of conversion symptoms, although a recent study finds that this clinical sign does not reliably discriminate between conversion disorder and organic disease.82
Summary
Finally, evaluating a young person with stroke symptoms necessarily requires a broader differential than a similar presentation in an older population. The workup should include unique elements of history, examination, and diagnostic testing to rapidly diagnose and treat stroke syndromes common in the younger population. The history must include family history of thrombotic or vascular processes, as well as the patient's past history of migraine, prior thrombotic events, or diseases associated with risk (i.e., systemic lupus erythematosis), and medications (i.e., oral contraceptives, warfarin) or illicit drug use. Further, the history of the current event should query whether pain in the head or neck was present, and the temporal course of the symptoms.
The diagnostic evaluation must include a good neurologic examination with emphasis on pupils, cranial nerves, cerebellar function, as well as extremity motor and sensory findings. A GCS is appropriate on all patients with any decrease in level of responsiveness as a way of following neurologic status over time.
Routine laboratory studies should be drawn, including coagulation studies, and an electrocardiogram and cardiac monitoring should occur (in part, to rule out atrial fibrillation as a source of emboli). Concern about an embolic source of stroke in a young person should lead to a transesophageal echocardiography to evaluate intracardiac clots, as well as right to left shunts, including the presence of a PFO. A noncontrast CT scan is the appropriate first imaging test, and will reliably exclude intracerebral hemorrhage. Unfortunately, CT is notoriously unhelpful in cervical artery dissection and cerebral venous thrombosis. When there is concern for these two diagnoses, MRI with arterial and with or without venous phase contrast is appropriate, or at least helical CT with contrast.
References
1. Kristensen B, Malm J, Carlberg B, et al. Epidemiology and etiology of ischemic stroke in young adults aged 18 to 44 years in northern Sweden. Stroke 1997;28:1702-1709.
2. Lee TH, Hsu WC, Chen CJ, et al. Etiology study of young ischemic stroke in Taiwan. Stroke 2002;33:1950-1955.
3. Leys D, Bandu L, Henon H, et al. Clinical outcome in 287 consecutive young adults (15–45 years) with ischemic stroke. Neurology 2002;59:26-33.
4. Jacobs BS, Doden-Albala B, Lin IF, et al. Stroke in the young in the northern Manhattan Stroke Study. Stroke 2002;33:2789-2793.
5. Bogousslavsky J, Pierre P. Ischemic stroke in patients under age 45. Neurol Clin 1992;10:113-124.
6. Hagan PT, Scholtz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc 1984;59:17-2.
7. Overell JR, Bone I, Lees KR. Interarterial septal abnormalities and stroke: a meta-analysis of case-control studies. Neurology 2000;55:1172-1179, 2000.
8. Wu LA, Malouf JF, Dearami MD, et al. Patent foramen ovale in cryptogenic stroke. Arch Intern Med 2000;164:950-956.
9. Meissner I, Whinsnant JP, Khandheria BK, et al. Prevalence of potential risk factors for stroke assessed by transesophageal echocardiography and carotid ultrasonography: the SPARC study: Stroke Prevention: Assessment of Risk in a community. Mayo Clin Proc 1999;74:862-869.
10. Loscalzo J. Paradoxical embolism: Clinical presentation, diagnostic strategies, and therapeutic options. Am Heart J 1986;112:141-145.
11. Cabanes L, Mas JL, Cohen A, et al. Atrial septal aneurysm and patent foramen ovale as risk factors for cryptogenic stroke in patients less than 55 years of age: A study using transesophageal echocardiography. Stroke 1993;24: 1865-1873.
12. Agmon Y, Khandheria BK, Meissner I, et al. Frequency of atrial septal aneurysms in patients with cerebral ischemic events. Circulation 1999;99: 1942-1944.
13. Hanley PC, Tajik AJ, Hynes JK, et al. Diagnosis and classification of atrial septal aneurism by two-dimensional echocardiography: report of 80 consecutive cases. J Am Coll Cardiol 1985;6:1370-1382.
14. Mugge A, Daniel WG, Angermann D, et al. Atrial septal aneurysm in adult patients: A multicenter study using transthoracic and transesophageal echocardiography. Circulation 1995;91:2785-2792.
15. Rahmatullah AF, Rahko PS, Stein JH. Transesophageal echocardiography for the evaluation and management of patients with cerebral ischemia. Clin Cardiol 1999;22:391-396.
16. Homma S, Sacco MD. Patent foramen ovale and stroke. Circulation 2005; 112:1063-1072.
17. Mas J-L, Arquizan C, Lamy C, et al. Recurrent cerebrovascular events associated with patent foramen ovale, atrial septal aneurysm, or both. N Engl J Med 2001;345:1740-1746.
18. Bogousslavsky J, Barazi S, Jeanrenaud X, et al. Stroke recurrence in patients with patent foramen ovale: the Lausanne Study. Neurology 1996;46: 1301-1305.
19. Kizer JR, Devereux RB. Patent foramen ovale in young adults with unexplained stroke. N Engl J Med 2005;355:2361-2372.
20. Schievink WI. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med 2001;344:898-906.
21. Thani B, Munshi SK, Dawson SL, et al. Carotid and vertebral artery dissection syndromes. Postgrad Med J 2005;81:383-388.
22. Caplan LR, Biousse V. Cervicocranial arterial dissections. J Neuro-Ophthalmol 2004;24:299-305.
23. Benninger DH, Georgiadis D, Kremer C, et al. Mechanism of ischemic infarct in spontaneous carotid dissection. Stroke 2004;35:482-485.
24. Lucas C, Moulin T, Deplanque D, et al. Stroke pattern of internal carotid artery dissection in 40 patients. Stroke 1998;29:2646-2648.
25. Rubinstein SM, Peerdeman SM, van Tulder MW, et al. A systematic review of the risk factors for cervical artery dissection. Stroke 2005;36:1575-1580.
26. Smith WS, Johnston SC, Skalabrin EJ, et al. Spinal manipulative therapy is an independent risk factor for vertebral artery dissection. Neurology 2003; 60:1424-1428.
27. Ernst E. Manipulation of the cervical spine: A systematic review of case reports of serious adverse events, 1995-2001. MJA 2002;176:376-380.
28. McIntosh A, Hungs M, Kostanian V, et al. Carotid artery dissection and middle cerebral artery stroke following methamphetamine use. Neurology 2006; 67:2259-2260.
29. Lee VH, Brown RD, Mandrekar JN, et al. Incidence and outcome of cervical artery dissection. Neurology 2006;67:1809-1812.
30. Arnold M, Cumurciuc R, Stapf C, et al. Pain as the only symptom of cervical artery dissection. J Neurol Neurosurg Psychiatry 2006;77:1021-1024.
31. Arnold M, Bousser MG, Fahrni G, et al. Vertebral artery dissection: presenting findings and predictors of outcome. Stroke 2006;37:2499-2503.
32. Phan T, Huston J, Bernstein MA, et al. Contrast-enhanced magnetic resonance angiography of the cervical vessels. Stroke 2001;32:2282-2286.
33. Quint DJ. Indications for emergent MRI of the central nervous system. JAMA 2000;283:853-855.
34. Schievink WI. The treatment of spontaneous carotid and vertebral artery dissections. Curr Opin Cardiol 2000;15:316-321.
35. Arnold M, Nedeltchev K, Sturzenegger M, et al. Thrombolysis in patients with acute stroke caused by cervical artery dissection. Arch Neurol 2002;59: 549-553.
36. Georgiadis D, Lanczik O, Schwab S, et al. IV thrombolysis in patients with acute stroke due to spontaneous carotid dissection. Neurology 2005;64: 1612-1614.
37. Dentali F, Crowther M, Ageno W. Thrombophilic abnormalities, oral contraceptives and risk of cerebral vein thrombosis: A meta-analysis. Blood 2006; 107:2766-2773.
38. Crassard I, Bousser M. Cerebral venous thrombosis. J Neuro-Ophthalmol 2004;24:156-163.
39. Lanska DJ, Kryscio RJ. Risk factors for peripartum and postpartum stroke and intracranial venous thrombosis. Stroke 2001;31:1274-1282.
40. Vandenbroucke JP, Rosing J, Bloemenkamp K, et al. Oral contraceptives and the risk of venous thrombosis. N Engl J Med 2001;344:1527-1535.
41. DiNubile MJ. Septic thrombosis of cavernous sinuses. Arch Neurol 1988;45: 567-572.
42. Cumuriciuc F, Crassard I, Sarov M, et al. Headache as the only neurological sign of cerebral venous thrombosis: A series of 17 cases. J Neurol Neurosurg Psychiatry 2005;76:1084-1087.
43. Stam J. Thrombosis of the cerebral veins and sinuses. N Engl J Med 2005; 352:1791-1798.
44. Ameri A, Bousser MG. Cerebral venous thrombosis. Neurol Clin 1992;10: 87-111.
45. Ehtisham A, Stern B. Cerebral venous thrombosis: A review. Neurologist 2006;12:32-38.
46. Smith R, Hourihan MD. Rational imaging: Investigating suspected cerebral venous thrombosis. BMJ 2007;334:794-795.
47. Wetzel SG, Kirsch E, Stock KW, et al. Cerebral veins: Comparative study of CT venography with intraarterial digital subtraction angiography. Am J Neuroradiol 1999;20:249-255.
48. Khandelwal N, Agarwa A, Kochhar R. Comparison of CT venography with MR venography in sinovenous thrombosis. Am J Roentgen 2006;187: 1737-1743.
49. De Bruijin SFTM, Stam J. Randomized, placebo-controlled trial of anticoagulant treatment with low-molecular-weight heparin for cerebral sinus thrombosis. Stroke 1999;30:484-488.
50. Einhaupl KM, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet 1991;338:597-600.
51. Ferro JM, Canhao MD, Stam MD. Prognosis of cerebral vein and dural sinus thrombosis: Results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke 2004;35:664-670.
52. Bousser MG, Chiras J, Bories J. Cerebral venous thrombosis-a review of 38 cases. Stroke 1985;16;199-213.
53. Bushnell CD, Siddiqu A, Goldstein LB. Improving patient selection for coagulopathic testing in the setting of acute ischemic stroke. Neurology 2001;57:1333-1335.
54. Busnell CD, Goldstein lB. Diagnostic testing for coagulopathies in patents with ischemic stroke. Stroke 2000;31:3067-3078.
55. Kahn MJ. Hypercoagulability as a cause of stroke in adults. South Med J 2003;96:350-353.
56. Bushnell CD, Goldstein LB. Physician knowledge and practices in the evaluation of coagulopathies in stroke patients. Stroke 2002;33:948-953.
57. Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta-analysis. JAMA 2000;284:72-78.
58. Chan WS, Ray J, Wai, EK, et al. Risk of stroke in women exposed to low-dose oral contraceptives. Arch Intern Med 2004;164:741-747.
59. Witlin AG, Mattar F, Sibai BM. Postpartum stroke: A twenty-year experience. Am J Obstet Gynecol 2000;183:1:83-88.
60. Kittner SJ, Stern BJ, Feeser BR, et al. Pregnancy and the risk of stroke. N Eng J of Med 1996;335:768-774.
61. James A H, Bushnell CD, Jamison MG. Incidence and risk factors for stroke in pregnancy and the puerperium. Obstet Genecol 2005;106:509-516.
62. Qureshi AI, Tuhrim S, Broderick JP, et al. Spontaneous intracerebral hemorrhage. N Engl J Med 2001;344:1450-1460.
63. Badjatia N, Rosand J. Intracerebral hemorrhage. Neurologist 2005;11: 311-324.
64. Ariesen MJ, Claus SP, Rinkel GJE, et al. Risk factors for intracerebral hemorrhage in the general population. Stroke 2003;34:2060-2066.
65. Westover AN, McBride S, Haley RW. Stroke in young adults who abuse amphetamines or cocaine. Arch Gen Psychiatry 2007;64:495-502.
66. Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med 2000;343:1826-1832.
67. Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology 2007;68:116-121.
68. Thompson KM, Gerlach SY, Jorn KS, et al. Advances in the care of patients with intracerebral hemorrhage. Mayo Clin Proc 2007;82:987-990.
69. Bateman BT, Schumacher HC, Bushnell CD, et al. Intracerebral hemorrhage in pregnancy. Neurology 2006;67:424-429.
70. Juvela S, Kase CS. Advances in intracerebral hemorrhage management. Stroke 2006;37:301-304.
71. Manno EM, Atkinson JLD, Fulgham JR, et al. Emerging medical and surgical management strategies in the evaluation and treatment of intracerebral hemorrhage. Mayo Clin Proc 2005;80:420-433.
72. Aguilar MI, Hart RG, Kase CS, et al. Treatment of Warfarin-associated intracerebral hemorrhage: Literature review and expert opinion. Mayo Clin Proc 2007;82:82-92.
73. Mendelow AD, Unterberg A. Surgical treatment of intracerebral hemorrhage. Curr Opin Crit Care 2007;13:169-174.
74. Qureshi AI, Janssen RS, Karon JM, et al. Human immunodeficiency virus infection and stroke in young people. Arcg Neurol 1997;54:1150-1153.
75. Cole JW, Pinto AN, Hebel JR, et al. Acquired immunodeficiency syndrome and the risk of stroke. Stroke 2004;35:51-56.
76. Nogueeras C, Sala M, Sasal M, et al. Recurrent stroke as a manifestation of primary angiitis of the central nervous system in a patient infected with human immunodeficiency virus. Arch Neurol 2002;59:468-473.
77. Cestari DM, Weine DM, Panageas KS, et al. Stroke in patients with cancer: incidence and etiology. Neurology 2004;62:2025-2030.
78. Wang WC. The pathophysiology, prevent, and treatment of stroke in sickle cell disease. Curr Opin Hematol 2007;14:191-197.
79. Golomb MR. Sickle cell trait is a risk factor for early stroke. Arch Neurol 2005;62:1778-1779.
80. Milhaud D, Bogousslavsky J, van Melle G, et al. Ischemic stroke and active migraine. Neurology 2001;57:1805-1811.
81. Diener HC, Kurth T, Dodick D. Patent foramen ovale, stroke, and cardiovascular disease in migraine. Curr Opin Neurol 2007;20:310-319.
82. Stone J, Smyth R, Carson A, et al. La belle indifference in conversion symptoms and hysteria: systematic review. Brit J Psychiatry 2006;188:204-209.
While stroke is most often a result of progressive atherosclerotic cerebrovascular disease, occurring with increasing frequency as the population ages, there are less common causes of stroke that mainly affect younger populations.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.