Emergency Department Care of Adult Cystic Fibrosis Patients
Emergency Department Care of Adult Cystic Fibrosis Patients
Authors: Lisa Freeman Grossheim, MD, FACEP, Assistant Professor, Department of Emergency Medicine, University of Texas Medical School at Houston; and Keith S. Gates, MD, EMT-P, Resident, Department of Emergency Medicine, University of Texas Medical School at Houston.
Peer Reviewer: Charles Emerman, MD, Cleveland Clinic, MetroHealth Medical Center, Cleveland, OH.
With advances in medical science, patients with serious congenital diseases are living into adulthood. A previous issue of Emergency Medicine Reports dealt with adults with congenital heart disease. This issue deals with another group of survivors, those who have survived with cystic fibrosis. Where once patients died in infancy, patients now live into their 40s. Although many of the standard treatments for COPD apply to cystic fibrosis, there are specific differences in management, which this monograph highlights.
Sandra M. Schneider, MD, FACEP, Editor
Introduction
A 19-year-old Caucasian male who was diagnosed with cystic fibrosis as a child presents to the emergency department in respiratory distress. His parents are at his bedside. The patient is thin, pale, chronically ill-appearing, and is in moderate distress with coughing and mild wheezing. His blood pressure is 110/60 mm Hg, heart rate 120, respiratory rate 48, temperature 99.5°F, and oxygen saturation 88% on 15 liters oxygen. The patient tells you that he was just discharged from the hospital last week and does not want to be readmitted or intubated. He says he is tired of being sick. His distraught parents are at his bedside and are asking you to do everything possible to help their son. What can you do? What should you do?
Overview
'The child will soon die whose forehead tastes salty when kissed," appeared in an 1857 Almanac of Children's Songs and Games from Switzerland 100 years before the sweat test was first used in the 1950s to help diagnose cystic fibrosis (CF), a disease that is characterized by abnormal salt transport, and, in previous years, death in infancy. Cystic fibrosis (CF) was first described as a disease entity in 1938.1,2
The important developments of the past few decades in cystic fibrosis care have led to the tremendous growth of the adult CF population. Cystic fibrosis has classically been defined as a pediatric disease since, at the time of discovery, it was invariably fatal in infancy, and in the 1950s median survival was 6 months. The median predicted survival was 16 years in 1970s. For patients born in the 1990s, median survival is predicted to be over 40 years.3,4 More than 36% of patients in the U.S. Cystic Fibrosis Registry are 18 years of age or older.5 Advances in medications and overall aggressive approach to the disease have led to this. Improved patient outcomes have been associated with comprehensive CF care centers and a multidisciplinary approach to patient care.1
Genetics and Epidemiology
Although there are reports dating from the 1650s of infants who likely had CF, the first report identifying the disease as a distinct clinical entity was published in 1938 by Dorothy Hansine Andersen, a pathologist at the Babies' Hospital in New York.2 She reported mucus plugging of the ducts of the pancreas in infants dying of malnutrition. She also first hypothesized that CF is a recessive disease. During the 1948 heat wave in New York, Paul di Sant'Agnese recognized that many of the infants presenting to the emergency department with heat-related illness and hyponatremic dehydration had CF. In subsequent summers, he determined that these patients had abnormally high sodium and chloride content in their sweat. In 1953, Dr. di Sant'Agnese and a colleague developed the sweat test, which measures for abnormal chloride levels in perspiration and, in 1959, standardization of the sweat test established this as the gold standard of CF diagnosis. In 1983, chloride transport was identified as the basic physiologic CF defect, accompanied by increased sodium reabsorption. In 1989, the discovery of the CF gene demonstrated the basic defect to be in a cAMP-regulated chloride channel.6,7
The CF trait results from a mutation of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) found on chromosome 7. Most homozygotes for the disease have the classic triad of chronic pulmonary disease, malabsorption secondary to pancreatic insufficiency, and elevated concentration of sweat electrolytes. Although this disease affects multiple organ systems and varies greatly in severity and progression, most morbidity and over 90% of mortality results from chronic lung disease.3
Cystic fibrosis is the most common lethal inherited disease among Caucasians in the United States with an incidence of 1 in 2000-3000 whites affected. About 1 in 20-25 whites carry mutations of the CFTR gene. African Americans are affected at a rate of 1 in 15,000 and Asian Americans at 1 in 31,000. All together, approximately 30,000 children and adults have CF in the United States. Cystic fibrosis is diagnosed in males and females equally. For unclear reasons, males tend to have a longer life expectancy than females.1
Pathophysiology
The CFTR is located in the apical membrane of epithelium in the pulmonary airways, pancreatic duct, intestine, biliary ducts, and the sweat glands. The CFTR protein crosses the membranes and acts as a channel connecting cytoplasm to the surrounding fluid. This channel is the primary means for controlling the movement of chloride across the cell membrane. The CF phenotype is expressed when the patient inherits two copies of the defective gene that encodes the CFTR protein. The most common mutation associated with CF is the deletion of three base pairs that code for phenylalanine at position F508 in the 1480 amino acid sequence of the CFTR protein.8 Once the deletion occurs, the mutant protein does not fold properly, so it is degraded. The exocrine glands are primarily affected because a lack of CFTR gene encoded transport proteins leads to the trapping of chloride inside the cells of the glands and on the surface of the skin. Without enough functional copies of the CFTR protein in their cell membranes, epithelial cells cannot pump enough water into the mucus and other products they secrete. Therefore, the secretions are too dry, thick, and sticky and they obstruct the small airways of the lungs and ducts of various organs. The abnormally viscous mucous secretions create a nutrient rich environment that is protected from the host immune system. This leads to chronic infection, inflammation, or both and eventually leads to tissue destruction and remodeling, resulting in bronchiectasis.8 Bronchiectasis refers to localized, irreversible dilatation of bronchi. Involved bronchi are dilated, inflamed, and easily collapsible, resulting in airflow obstruction and impaired clearance of secretions.
 |
Diagnosis
Although diagnosis through DNA analysis is becoming a standard of care in CF, the sweat test is still the diagnostic gold standard. It is the only method of diagnosis recognized by the Cystic Fibrosis Foundation as an adequate test for definitive diagnosis of CF.5 The test involves collection of sweat by pilocarpine iontophoresis coupled with chemical determination of the chloride concentration.9,10 At least 50 mL of sweat must be collected in a 30-minute period. Using a chloride threshold of 60 mEq/L in sweat appears to distinguish nearly all adults with CF from those with other lung conditions.11 However, normal values of sweat chloride do rise with age, so higher levels may be considered normal in some patients. One to two percent of patients have clinical features consistent with CF but have normal sweat chloride levels. These patients can be diagnosed by detecting mutations in the CF gene.6
Clinical Presentation
CF runs a highly varied course, ranging from death due to complications from meconium ileus in the first days of life or death from severe respiratory tract problems within the first few months of life to minimally symptomatic course for 10 to 20 years and extended survival. An increasing number of patients with CF live into the sixth and seventh decades of life.6,7,12
Cough is usually the earliest symptom and is usually worse at night. At first it is intermittent and occurs with what appears to be an acute respiratory illness. The coughing is sometimes accompanied by wheezing, especially in infants and young children. Episodes of coughing persist longer than expected for a routine respiratory illness and occur more frequently with time. As the disease progresses, the cough becomes productive of thick purulent sputum. Symptoms consistent with bronchitis may occur for several years before diagnosis. Eventually the exacerbations of productive cough are accompanied by dyspnea, anorexia, and weight loss. Physical findings depend on the stage of the disease. At first crackles are intermittent and occur with exacerbations. Lung sounds may be decreased because of hyperinflation. As the disease progresses, rales and rhonchi are common and continuous.8 The majority of adults with CF die of respiratory failure.
Patients who have not yet been diagnosed with CF may initially be seen at any age and can present with a variety of complaints. Many symptoms can mimic those found in a variety of other diseases. Failure to thrive and a history of chronic respiratory, and/or gastrointestinal problems are the most common symptoms in children. A child may have persistent cough or recurrent pneumonia and atelectasis. Atypical asthmatics with digital clubbing (which occurs in nearly all patients with CF), bronchiectasis, or cough producing sputum may potentially have CF. Undiagnosed cystic fibrosis patients may have been seen in the emergency department or primary care physician's office multiple times for respiratory or GI complaints, yet the diagnosis remains elusive. CF won't be diagnosed in patients with mild disease or older patients unless someone puts the pieces together and refers the patient for testing. This is an important role for the emergency physician.
Patients diagnosed with CF as adults usually present with chronic respiratory problems. As a group, they have milder lung disease, less Pseudomonal infection, and are more likely to be pancreatic-sufficient than patients diagnosed at an earlier age.13-15 Adult patients come to attention with atypical presentations such as chronic/recurrent pancreatitis, recurrent pneumonia, or bronchitis.3,16-18
A newborn may have a meconium ileus while older children and adults can present with frequent passage of pale, bulky, loose, and excessively foul-smelling stool that is characteristic of CF. Young patients are often misdiagnosed as having milk allergy or chronic diarrhea and may have tried multiple different formula preparations. Edema and hyponatremia may develop in children with CF, especially those taking a soy protein formula. Bleeding problems resulting from vitamin K malabsorption may be seen. Many patients presenting with less severe symptoms may not require emergency treatment at all. These patients, however, must be referred for further diagnostic evaluation if the suspicion of CF is high.
 |
 |
Pulmonary Disease. Changes consistent with airway obstruction appear first in the small airways along with evidence of airway hyperactivity. Arterial oxygenation decreases with time. As the disease progresses, untreated hypoxemia and progressive loss of functional lung may produce pulmonary artery hypertension and right ventricular failure (cor pulmonale). Hypercarbia and chronic respiratory acidosis are apparent in late stage disease. Respiratory failure becomes increasingly difficult to manage at this point.
The lung function in adults with CF is highly variable. FEV1 expressed as the percentage predicted of a healthy nonsmoking reference population is accepted as the single most useful objective measure of pulmonary status.19 Thirty-six percent of adults with CF have normal or mild lung dysfunction (FEV1 > 70% predicted). Thirty-nine percent have moderate dysfunction (FEV1 < 40% to 69%). The remainder have severe dysfunction (FEV1 < 40% predicted).3
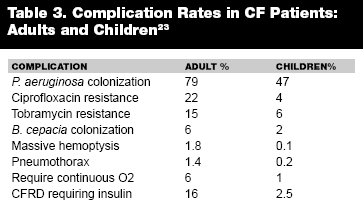
The lungs of CF patients are colonized and infected by bacteria from an early age. Pseudomonas aeruginosa is the most common pathogen, infecting most of the CF population.20 In fact, persistent bacterial pulmonary infection, especially with Pseudomonas aeruginosa, is the hallmark of CF. The mucoid strains of Pseudomonas are associated with more rapid clinical deterioration. Although mucoid P. aeruginosa is occasionally cultured from patients with other lung diseases, its presence in the sputum should immediately alert the physician to the possibility of cystic fibrosis. Other common pathogens encountered include S. aureus, H. influenzae, Stenotrophomonas maltophilia, Burkholderia cepacia, and non-tuberculous mycobacteria.21 As the disease progresses, infection with antibiotic resistant bacteria and concomitant infection with more than one organism are likely.8 Burkholderia cepacia is a multiple resistant gram negative organism that carries the worst prognosis. Some patients who develop a sepsis-like syndrome with severe necrotizing pneumonia called cepacia syndrome have rapid deterioration and death.1
The most frequent fungal infection seen is Candida, affecting 50-70% of patients. Aspergillus is isolated from 25% of patients and can be associated with significant allergic manifestations such as wheezing, pulmonary infiltrates, and worsening of bronchiectasis and fibrosis.22 More than 50% patients have antibodies to Aspergillus fumigatus, but only a small percentage develop allergic bronchopulmonary aspergillosis. This is characterized by rusty brown sputum plugs.8
These organisms, which often spread among individuals with CF, thrive in the altered mucus in the small airways of the lungs. This mucus encourages the development of bacterial microenvironments called biofilms that are difficult for immune cells and antibiotics to penetrate. CF disease progresses from bronchiolitis at a young age to bronchitis and eventually bronchiectasis as a consequence of persistent obstruction and inflammation. Bronchiectatic cysts are prominent in 50% of lungs in end stage patients and may contribute to the 3-19% reported incidence of pneumothorax.1,8
In general, adults with CF have more severe pulmonary disease than children. Adults are at higher risk for serious complications such as pneumothorax and massive hemoptysis.
Chronic airway infection leads to significant, persistent neutrophilic inflammatory response that destroys small airways, leading to bronchiectasis. Angiogenesis in areas of intense inflammation predisposes patients to hemoptysis, which occurs in about 3% of adult patients per year. Hemoptysis, which usually presents as blood-streaked sputum, becomes more common as bronchiectasis develops. Significant hemoptysis is 30-60 cc of blood and is due to the erosion of an area of local bronchial infection or bronchiectasis compromising a bronchial vessel. Hemoptysis is usually self-limited, but embolization or lobectomy may be required in severe cases.1 Hemoptysis may require supplemental vitamin K if the prothrombin time is prolonged due to inadequate absorption.24
Pneumothorax is a well-known complication whose incidence increases with age. Sixteen to 20% of CF adults have a pneumothorax at some point, often as the result of rupture of a subpleural bleb. The patient usually presents with chest pain, dyspnea, and hemoptysis. CF patients with a pneumothorax of greater than 10% should be treated with a tube thoracostomy as 30% of these patients are reported to experience a tension pneumothorax.1,8
Additional diagnoses such as asthma, allergic bronchopulmonary aspergillosis, sinus disease, and gastroesophageal reflux should be considered in patients whose clinical course of respiratory decompensation or response to treatment are atypical for CF.
Loss of CFTR function also affects the upper airway epithelium, so chronic rhinitis is common. The sinuses are universally involved but acute/chronic sinusitis is uncommon.
Cardiac Disease. Patients with CF who have moderately severe pulmonary insufficiency and some degree of hypoxia will eventually develop right ventricular hypertrophy secondary to pulmonary hypertension (cor pulmonale). Increased hypoxia during an exacerbation of pulmonary symptoms in such patients may precipitate an episode of congestive heart failure. In addition to cyanosis, tachypnea, and tachycardia, other associated symptoms may include an enlarged, tender liver and ascites.25
Gastrointestinal Disease. Failure to secrete enough chloride and fluid in the intestine leads to reduced water content of the fecal stream, which results in meconium ileus in many infants with CF. The abnormal intestinal mucus in CF patients leads to a decrease in mobility that, combined with a decreased amount of abnormal pancreatic and biliary secretions, results in a dry, thick stool that cannot pass from the terminal ileum to the cecum. This may cause recurrent abdominal pain, obstruction, volvulus, or intussusception. In older patients, distal intestinal obstruction syndrome or "meconium ileus equivalent" can produce intermittent recurrent episodes of partial small bowel obstruction in 15% of patients and may lead to complete obstruction.1 Distal intestinal obstruction syndrome can be relieved with intestinal "flushes" with a balanced salt solution of 1-2 liters instilled into the stomach. Rectal prolapse may be seen in adults and can usually be reduced voluntarily by using abdominal, perianal, and gluteal muscles.6
Gastroesophageal reflux disease (GERD) is common in CF patients, and is seen in more than 20%.26 Many factors contribute, such as head down position for airway clearance, medications that decrease lower esophageal sphincter tone, and hyperexpansion of the lung that flattens the diaphragm and impairs the physiologic sphincter.27
Destruction and loss of pancreas function occurs at birth or in early infancy. Exocrine pancreas disease affects most CF patients; 91% are on pancreatic replacement therapy.27,28 Obstruction of the ducts, loss of acinar cells, and pancreatic enzyme deficiency leads to malabsorption of protein, fat, and fat-soluble vitamins. This leads to bulky, foul-smelling stool and weight loss.8 Abnormal CFTR function in the ducts of the pancreas causes a decreased volume of secretions with reduced bicarbonate concentration. Autoactivation of retained digestive proenzymes leads to destruction of pancreatic tissue. Consequently, absorption of fat soluble vitamins A,D,E, and K is reduced. The presence of pancreatic insufficiency portends a worse overall prognosis.
Symptoms of pancreatitis occur in a small percentage of adolescents and adults, especially in those who have retained pancreatic function. There is a strong association between idiopathic pancreatitis and having one CFTR mutation.29 Although the Islets of Langerhans are relatively spared, destruction of the pancreas can cause endocrine pancreatic dysfunction leading to diabetes. This rarely develops before the age of 10 and usually manifests after the second decade of life. The prevalence of CF-related diabetes increases with age from 9% (age 5-9) to 43% for patients over 30 years of age.30 Hyperglycemia can occur at any age but is generally a problem of the second and third decades of life.31 Diabetic ketoacidosis is rare in CF-related diabetes. Most patients with sustained hyperglycemia require insulin, but oral agents may work in some patients.
Liver disease is the second most common cause of death in CF patients.28 Many CF patients have some form of liver or biliary disease.8 This may manifest as elevated transaminases, hepatosteatosis, or gall stones. Frank liver disease with cirrhosis and liver failure can occur in childhood and is progressive. As patients are living longer, chronic obstruction of the ducts may lead to liver damage and biliary cirrhosis. Hematemesis is a severe complication that may develop from esophageal varices due to portal hypertension. The severity of liver disease varies widely from mild elevation of alkaline phosphatase in many patients to hepatomegaly and persistently elevated liver enzymes to jaundice, ascites, and edema in severe cases. However, few patients develop clinical cirrhosis. Thirty percent of adult CF patients have a hypoplastic, poorly functioning gallbladder and may develop gallstones.1,3
There is an increased risk of Crohn's disease by 12-fold in CF patients over the general population as well as a 6-fold increase risk of malignancy.32,33
Genitourinary Disease. Atrophy of the Wolffian duct structures is almost universal in CF patients. Ninety-five percent of young men with CF are infertile because of bilateral absence of the vas deferens, abnormalities of the seminal vesicle, or both.34 It is believed that the vas deferens becomes occluded during gestation and is reabsorbed. Spermatogenesis is retained, however, and retrieval of sperm for in vitro fertilization can be performed. Females with CF have decreased fertility because of thick secretions at the cervical os and poor nutritional status.34-36
Emergency Department Evaluation and Treatment
Radiographs. Hyperinflation is often the earliest change seen on chest radiograph. Subsequent peribronchial thickening creates peribronchial cuffing. As the disease progresses, mucus impaction and bronchiectasis are seen as well as variable amounts of fluffy infiltrates. The right upper lobe is usually the first and most severely involved. Widespread bronchiectasis may be seen on CT before it appears on plain chest radiographs.8
A chest radiograph is indicated for patients with suspicion for an acute complication, such as pneumonia or pneumothorax. Comparison to old films is important to note acute changes. Chest CT is indicated for patients presenting with complications such as loculated pleural effusions, lung abscess, or other potentially surgical problems.
Laboratory evaluation is complaint-specific may include complete blood count, basic metabolic panel, transaminases, coagulation studies, and arterial blood gases.
The cornerstones of emergency treatment in CF are similar in adults and children. This includes oxygen, airway support, antibiotic treatment (acute, chronic, or suppressive), airway clearance, mucolytic therapy, anti-inflammatory agents, and bronchodilators.
Oxygen. Supplemental oxygen should be used as needed to maintain adequate oxygenation in the acute setting. Supplemental oxygen in accordance with the guidelines established for chronic COPD is recommended.37,38 The most important chronic therapy for the prevention of pulmonary hypertension is supplemental oxygen.
Antibiotics. Pulmonary exacerbations are common in adults with CF and usually are associated with bacterial infections. There is evidence that early, aggressive use of antibiotics in decompensated CF patients produces better results than delaying the administration of antibiotics until symptoms are well developed or advanced.39 The choice of antibiotics and the use of single or combined therapy are controversial areas in the treatment of respiratory infection in CF. A recent Cochrane review investigated single vs. combination intravenous antibiotic therapy for CF patients. The results were inconclusive due to multiple trials, variable antibiotic choices, and significant methodological issues.40
Although P. aeruginosa is rarely eradicated once it becomes chronic, an important benefit is gained by decreasing the net bacterial load with intensive parenteral antibiotics. As the number of organisms decreases, airway inflammation is reduced, thus decreasing the airway destruction and the airway symptoms. Azithromycin has no direct killing effect against Pseudomonas but it can adversely affect Pseudomonas virulence factors and it is active against H. influenzae and S. aureus.41 There is evidence that the macrolides demonstrate acute inflammatory effects such as modulation of signaling pathways, inhibition of proinflammatory cytokines, limiting influx of neutrophils to the lung, mucus secretion, and altering the formation of the biofilm matrix.41-43
Long-term macrolide antibiotics effectively treat diffuse panbronchiolitis and produce clinical improvement in patients chronically infected with Pseudomonas. Azithromycin has been shown in 4 randomized controlled trials to improve lung function and reduce the frequency of pulmonary exacerbations,42,44-46 even prior to infection with Pseudomonas.
Antibiotics are selected on the basis of recent sputum cultures, if available, although this is not often the case in the emergency department. Therapy with fluoroquinolones is often used for mild to moderate exacerbations in adults. Two antipseudomonal antibiotics are used in combination (ie. Beta lactam and aminoglycoside) for the treatment of moderate to severe exacerbations.3 (See Table 4 for suggested antibiotic regimens.)
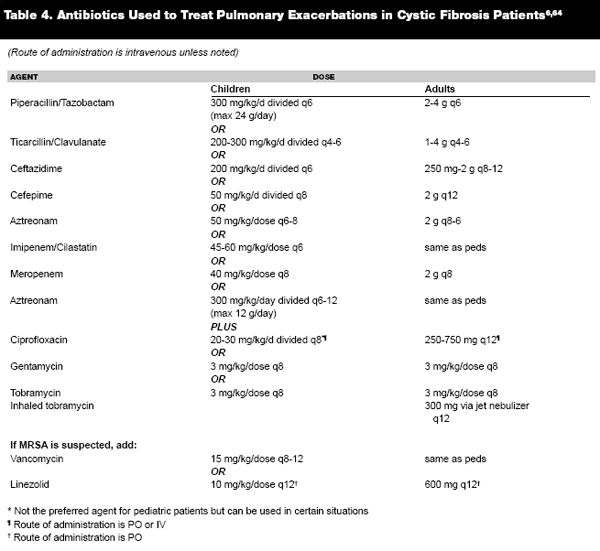 |
Aminoglycosides have been the mainstay of anti-Pseudomonas therapy for many years and may still have a role in some patients. A major advantage is the ability to monitor and adjust blood levels. Disadvantages include oto- and nephrotoxicity. A drug like gentamycin or tobramycin is usually paired with one of the penicillin derivatives or with ceftazidime.26 Clearance of aminoglycosides by the kidney is increased in CF. The re-quired doses are often two to three times higher than in non-CF patients.8
Inhalation antibiotics are attractive because high concentrations can be attained at the airway surface and in mucus, thereby minimizing systemic toxicity. Aerosolized high-dose tobramycin (TOBI) can reduce the density of P. aeruginosa, improve FEV1, and reduce length of hospitalization.47
Chronic suppressive antibiotic therapy is increasingly becoming a standard part of care.
Aerosolized tobramycin is the most thoroughly studied chronic suppressive therapy. In two large double blind placebo controlled trials, treatment with TOBI was found to produce significant improvement in pulmonary function, to decrease the density of P. aeruginosa in the sputum, and to decrease the number of days the patients were hospitalized.45 Chronic, continuous, low-dose azithromycin also improves lung function and reduces the frequency of exacerbations.48,49
CF patients require higher doses of antibiotics and shorter dosing intervals. In general, the highest recommended doses are given to achieve penetration into the respiratory secretions. Both total body clearance and volume of distribution are considerably greater for CF patients than other patients.50 In addition, large doses are needed to achieve therapeutic levels in the infected and mucus- or pus-filled endobronchial space. Longer courses of 2-4 weeks of antibiotics are often used.
Chest Physiotherapy. Excessive bronchial secretions contribute to the airway obstruction in CF, which leads to atelectasis and hyperinflation. For 40 years, chest physiotherapy (CPT) was the major airway clearance strategy in CF. The goal of CPT is to improve pulmonary status and prolong survival via removal of tenacious bronchial secretions, reduce airway resistance, and improve ventilation over the short term.51 Chest PT facilitates loosening and expectoration of mucus. CPT is often initiated in asymptomatic patients to try to slow the progression of the disease.
The most compelling argument for the use of postural drainage with chest percussion (based on the concept that cough clears mucus from large airways but chest vibrations are necessary to move secretions from the small airways where expiratory flow rates are low) comes from a study of older children with mild-moderate airflow limitation.52 When patients were receiving CPT on a regular basis, the only immediate effect was an increase in peak expiratory flow rate 30 minutes after therapy. However, after 3 weeks without CPT, both functional vital capacity (FVC) and flow rates were significantly reduced. A meta-analysis of 35 studies concluded that standard CPT increases sputum production and improves expiratory airflow (FEV1).39
A widely used device for chest compression therapy is called the vest. It is a chest wall compression and oscillation system that is composed of a fitted vest and oscillation system coupled to a pneumatic compressor. Therapy is delivered to the entire chest at the same time with the patient in a seated position. This allows for the administration of nebulized medications during therapy and also affords independence to the patient.3
CPT may be utilized as part of acute therapy in the emergency department. This may be performed by a respiratory therapist or trained family member.
Bronchodilators. The majority of CF patients have bronchial hyperreactivity/bronchospasm at least some of the time.42 Bronchodilators are a standard component of the therapeutic regimen, and most emergency physicians are very familiar with the use of these medications. They are often used in conjunction with CPT to facilitate airway clearance. There is some evidence that maintenance albuterol reverses the progressive downhill course in lung function in CF patients.53-55
Anticholinergic bronchodilators may be helpful in some patients with CF. Ipratropium may be more effective than beta agonists in adults with CF. Adults often have less bronchospasm, but more secretions than children. The airway of the adult CF patient may closely mimic that of the adult with chronic bronchitis and, therefore, may be more responsive to the effects of a parasympathomimetic agent. Combination therapy with a beta agonist may also be effective.56-58
Hypertonic Saline. The excessive absorption of salt from the airway lumen of CF patients carries water with it, dehydrating airway mucus secretions and depleting the volume of liquid on the airway surface. These changes disrupt the mucociliary mechanism, and the retained mucus becomes a nidus for chronic infection.59 Therefore it is hypothesized that the use of hypertonic saline should be helpful in CF patients.
Over the short term, hypertonic saline (HS) improves the transportability of sputum and hydration of the airway surface60,61 and mucociliary clearance and lung function62,63 in patients with CF. Treatment with hypertonic saline for one year had no significant effect on the rate of decline in lung function, but it was associated with a moderate yet sustained improvement in the level of lung function. More dramatic were the reductions in the number of exacerbations, antibiotic use for exacerbations, and absenteeism from school/work.59
In a double blind study of 164 patients 6 years old or older with stable CF, patients inhaled 7% saline or normal saline twice a day for 48 weeks. The hypertonic group had significantly higher functional vital capacity (FVC) (by 82 cc, 95% CI 12-153) and FEV1 (by 68 cc, 95% CI 3-132). In addition, the hypertonic saline group had fewer pulmonary exacerbations (relative reduction of 56%, p = 0.02) and a higher percentage of patients without exacerbations (76% vs 62%, p = 0.03).59
Hypertonic saline, usually preceded by a bronchodilator, is inexpensive, safe, and effective therapy for patients with CF and can safely be used as part of the treatment in the emergency department.
Mucoactive Therapies. The most characteristic feature of inflammation in the lung is the persistent infiltration of massive numbers of neutrophils into the airway. Although neutrophils help control infection, when present in excess, they may cause more harm than good.65 Neutrophils infiltrating the airways and degenerating are the major source of the DNA and filamentous actin that makes CF sputum so tenacious.66,67 Augmentation of DNA content is directly associated with an increase in mucus viscosity.68 Mucoactive therapies reduce viscosity and tenacity of the sputum via degradation of the excess DNA. The most widely used agent for this purpose is dornase alpha (Pulmozyme).51
DNase is produced naturally in humans. More than 50 years ago, it was shown that bovine pancreatic deoxyribonuclease I (DNase I), an enzyme that cleaves DNA, reduced the viscosity of lung secretions in vitro.69 It was approved for human use in 1958. The agent lost popularity over time due to severe pulmonary reactions, possibly allergic in etiology. Recombinant human DNase (also known as alpha-dornase or Pulmozyme,) was sequenced in 1990 and used in aerosolized form. It decreases the viscosity of CF sputum by catalyzing extracellular DNA into smaller fragments.70 This preparation has better tolerability, with the most common adverse effects being respiratory such as pharyngitis and hoarseness with minor allergic reactions.51 The drug can be started safely during an acute pulmonary exacerbation, as well as during a stable period.71
A study of rhDNase in patients 3-16 years old showed that one-third children had sustained improvement in spirometry greater than 20% over 1 year, but one-third deteriorated.72 The effects of rhDNase on lung function diminish over time. Another study showed that after stopping rhDNase, lung function dropped markedly below the initial baseline level.51 The concern is that rhDNase only effects a superficial removal of secretions while deeper down below the mucosal surface, tissue damage continues as before. The dose is 2.5 mg nebulized once or twice daily.51
Ballman, et al., compared nebulized rhDNase to hypertonic saline in a short-term study of 14 patients with mild to moderate CF. They demonstrated comparable short-term effects and noted that hypertonic saline was much cheaper. A longer trial with 48 patients demonstrated that rhDNase produced significantly greater improvement in FEV1 from baseline compared with hypertonic saline.73
There are no other well-validated alternative mucolytic agents available presently. IV acetylcysteine reduces the viscosity of sputum in vitro but can be very irritating to the upper airway and can cause bronchoconstriction.74 Further investigation is ongoing.
To the authors' knowledge, no randomized controlled studies exist to support the efficacy of nebulized DNase in the emergency department for acutely decompensated pulmonary disease in CF patients. However, it is a reasonable addition to acute therapy in a CF patient in whom intubation is an undesirable option.
Steroids. Oral corticosteroids have a wide range of anti-inflammatory effects, one of which is to prevent the conversion of neutrophils to the activated state. They also prevent the production of toxic metabolites.41 Two randomized controlled trials in CF patients with mild to moderate disease treated with 1 or 2 mg/kg prednisone on alternate days for a 2-4 year period showed improvements in lung function and reduction in frequency of pulmonary exacerbations compared to placebo.75,76
Short-term treatment (3 weeks) with daily corticosteroids in stable patients with severe obstructive disease showed no benefit,77 but patients with less severe disease showed some improvement.78 The 1 mg/kg group vs placebo for 4 years had benefit with respect to pulmonary function at the expense of growth impairment and bone density.79,80
The long-term adverse effects are well-described and include glucose intolerance, cataracts, and growth impairment. An increase in the rate of infection is not seen.76,81 Inhaled steroids have less systemic effects. Low doses of inhaled beclomethasone showed no effect on various markers of airway inflammation.82 Higher doses of inhaled steroids have shown promise in preliminary studies.83,84 Larger trials with long-term data are needed.
Advanced Airway Management. Hypercapnic and hypoxemic respiratory failure in CF is primarily due to progressive obstructive airway disease with alveolar hypoventilation with ventilation-perfusion mismatch. When a CF patient presents in respiratory failure, the management decisions become difficult. CF patients in general do not respond as well to and have more complications from mechanical ventilation compared to COPD patients.25 However, ventilatory assistance is effective in CF patients with acute respiratory failure caused by reversible insults,40 but produces few long-term benefits in patients with respiratory failure due to irreversible bronchiectasis.85 The difference between these two entities may not be easy to determine the emergency department setting. Aggressive ICU care for adults with CF who have respiratory failure as a consequence of progression of their disease can be beneficial.86 If an acute episode such as pneumonia or bronchospasm precipitates respiratory failure in a CF patient who had good pulmonary function before the episode, mechanical ventilation should be considered.25 Factors to consider when making the decision: the patient's level of activity and pulmonary function before the episode, the cause of the patient's decompensation, and the expectations of patient and his or her family. Ventilatory support is appropriate for a patient when lung transplant is planned.86 Consult with the patient's personal physician before electing not to intubate a patient.
Avoidance of endotracheal intubation and mechanical ventilation is desirable to limit possible airway complications of intubation, including nosocomial pneumonia and deleterious cardiovascular effects of positive-pressure ventilation such as decreased cardiac output or pneumothorax. Alternative ventilatory support in the form of BIPAP may be useful. Bilevel positive airway pressure (BIPAP) is a noninvasive mode of ventilation administered through a tight-fitting mask to assist spontaneously breathing patients. The BIPAP system regulates the pressure supplied to patients, delivering different pressures during inspiration and exhalation. As the patient initiates a breath, the system delivers air with a positive pressure gradient, thus increasing tidal volume and minute ventilation.87,88
BIPAP has been used in cystic fibrosis as a bridge to transplantation. It has been shown that the use of BIPAP is associated with improvement in ventilation and arterial blood gases as well as improvement in subjective symptoms such as headaches, activity level, and quality of sleep in CF patients with end-stage lung disease awaiting lung transplant.89,90 In a small study, Efrati, et al., demonstrated a significant improvement in survival of CF patients after lung transplant for patients who used BIPAP in the months before lung transplantation compared with those who did not use BIPAP. This improvement was thought to be due to improvement in respiratory muscle strength and nutritional status.87
Patient cooperation is crucial to the success of BIPAP. It is not recommended for patients with altered mental status, abnormal gag reflex, inability to protect airway, or inability to effectively clear respiratory secretions or protect their airway.
Heliox has been used with some success in asthmatic patients with severe airway obstruction.19,91 The literature describing the use of heliox in cystic fibrosis is very limited. It was used in conjunction with noninvasive high-frequency percussive ventilation in a 5-year old child with severe acute respiratory failure resulting from advanced cystic fibrosis lung disease. The patient had dramatic improvement and avoided intubation in that case.19 Another case report details the successful use of heliox in a teenage girl with severe acute respiratory compromise from cystic fibrosis.92 The mechanism of action is thought to be improvement of gas exchange by enhancing molecular diffusion and by favoring laminar flow throughout the upper and lower airways. Further study is needed, but this seems like a reasonable therapeutic alternative for an acutely decompensated cystic fibrosis patient.19,86,91
Promising Therapies. Over the last 50 years the Cystic Fibrosis Foundation has been at the forefront of research efforts for the development of potential CF therapies. CF, however, is still a complicated and uniformly fatal disease. Novel treatments can be divided into two basic categories: those aimed at correction of the CFTR dysfunction and those aimed at mitigating the effects of having a limited number of CFTR channels. Definitive cure for CF would involve correcting the deficit left by the CFTR gene mutation. Restoration of even 5-10% of airway epithelial cells could correct the electrolyte transport defect.8 There has been a significant amount of promising research in this direction, including gene therapy, activation of alternate chloride channels, and improving function of native CFTR.1
Ibuprofen can slow the effects of CF by reducing inflammation, but long-term use of this drug at the high doses required for improvement can negatively affect the kidney and gastrointestinal tract as well as other organs. The use of N-acetylcysteine as an alternate method to reduce inflammation is being investigated. High-dose oral N-acetylcysteine (NAC), a glutathione prodrug, modulates inflammation in cystic fibrosis. NAC has been tested with some success in patients with other inflammatory lung problems, including chronic bronchitis, chronic obstructive pulmonary disease, and idiopathic pulmonary fibrosis.93 Oral NAC treatment not only increased the amount of glutathione in circulating neutrophils, it also decreased the number of neutrophils and the levels of elastase and interleukin-8 in the airways. In 2006 the researchers began a 24-week placebo-controlled Phase II trial of NAC in cystic fibrosis patients to confirm these findings.93
In the terminal phases of the disease, lung transplantation often becomes necessary for individuals with CF as lung function and exercise tolerance declines. A pancreatic or liver transplant may be performed at the same time to alleviate liver disease and/or diabetes. Lung transplantation is considered when lung function approaches a point where it threatens survival or requires assistance from mechanical devices.
Disposition
The literature describing emergency department care of the acutely decompensated cystic fibrosis patient is sparse. Many pediatricians and pulmonologists work diligently to keep the patients out of the emergency department with care in the clinic and at home. If the patient needs to come to the ED, hospital admission is usually the goal.
The most important indications for admission would include need for intravenous antibiotics and/or advanced airway management. The disposition of the patient should include input from the patient's primary care physician or pulmonologist.
End-of-Life Issues
For patients who are not transplant candidates and have progressive respiratory failure, it may be difficult to decide whether to offer intubation. Potentially reversible aspects of the disease, short-term goals, and the wishes of the patient and the family are important to consider.86 In patients who are not transplantation candidates, the decision to proceed with mechanical ventilation should be undertaken with the understanding that there is a limited chance for a good outcome.94 For terminal patients in whom mechanical ventilation is not planned, the primary palliative care issue is the management of dyspnea. Morphine infusions have been successfully used in these cases to provide comfort care.94 Advanced directives are helpful in this situation and should be sought.
References
1. Strausbaugh SD, Davis PB. Cystic fibrosis: A review of epidemiology and pathobiology. Clin Chest Med 2007;28:279-288.
2. Anderson D. Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Child 1938;56:344-389.
3. Yankaskas JR, Marshall BC, Sufian B, et al. Cystic fibrosis adult care. Consensus conference report. Chest 2004;125:1S-39S.
4. Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax 1991;46:881-885.
5. Cystic Fibrosis Foundation website (www.cff.org). Accessed January 25, 2008.
6. Boucher RC, Knowles MR, et al. Cystic fibrosis; chapter 38. In: Mason RJ, Murray JF, eds. Murry and Nadel's Textbook of Respiratory Medicine, 4th ed. Philadelphia: Elsevier Saunders; 2005.
7. di Sant'Agnese PA, Davis PB. Cystic fibrosis in adults: 75 cases and a review o 232 cases in the literature. Am J Med 1979;66:121-132.
8. Walsh MJ. Cystic fibrosis. In: Goldman L, Ausiello D, eds. Cecil Medicine, 23rd ed. Philadelphia: Saunders Elsevier; 2008: 627-631.
9. Sweat Testing Sample Collection and Quantitative Analysis, Approved Guideline Document C34-A, Wayne PA, National Committee for Clinical Laboratory Standards, 1994.
10. LeGrys VA. Sweat testing for the diagnosis of cystic fibrosis: Practical considerations. J Pediatr 1996;129:892-897.
11. Davis PB, Del Rios, Muntz JA, et al. Sweat chloride concentrations in adults with pulmonary diseases. Am Rev Respir Dis 1983;128:34-37.
12. Stern RC, Boat TF, Doershuk CF, et al. Cystic fibrosis diagnosed after age 13: Twenty-five teenage and adult patients including three asymptomatic men. Ann Intern Med 1977;87:188-191.
13. Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: Current survival and population estimates to the year 2000. Thorax 1991;46:881-885.
14. Gan KH, Geus WP, Bakker W, et al. Genetic and clinical features of patients with cystic fibrosis diagnosed after the age of 16 years. Thorax 1995;50:1301-1304.
15. Widerman E, Millner L, Sexauer W, et al. Health status and sociodemographic characteristics of adults receiving a cystic fibrosis diagnosis after age 18 years. Chest 2000;118:427-433.
16. Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998;339: 653-658.
17. Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 1998;339:653-658.
18. Wang X, Moylan B, Leopold DA, et al. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA 2000;284:1814-1819.
19. Stucki P, Scalfaro P, de Halleux Q, et al. Successful management of severe respiratory failure combining heliox with noninvasive high-frequency percussive ventilation. Crit Car Med 2002;30:692-694.
20. Schidlow DV, Taussig LM, Knowles MR. Cystic Fibrosis Foundation consensus conference report on complications of cystic fibrosis. Pediatr Pulmonol 1993;15:187-198.
21. Burns JL, Emerson J, Stapp JR, et al. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis 1998;27: 158-163.
22. Greenberger PA. Allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol 2002;110:685-692.
23. Cystic fibrosis foundation. Patient registry 2000 annual data report. Bethesda, MD: Cystic fibrosis foundation, 2001.
24. Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet 1995;29:777-807.
25. Scanlin TF. Cystic fibrosis. In: Fleisher GR, Ludwig S. Textbook of Pediatric Emergency Medicine, 4th ed. Lippincott Williams and Wilkins; Philadelphia: 2000; 1087-1092.
26. Scott RB, O'Loughlin EV, Gall DG. Gastroesophageal reflux in patients with cystic fibrosis. J Pediatr 1985;106:223-227.
27. Ferry G, Klish W, Borowitz D, et al. Consensus conference for GI problems in cystic fibrosis. Bethesda, MD; June 1991.
28. Cystic Fibrosis Foundation Patient Registry 2005 Annual Data Report to the Center Directors. Bethesda, MD: Cystic Fibrosis Foundation 2006.
29. Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998;339; 653-658.
30. Moran A. Cystic fibrosis-related diabetes: An approach to diagnosis and management. Pediatr Diabetes 2000;1;41-48.
31. Robbins MK, Ontjes DA. Endocrine and renal disorders in cystic fibrosis. Cystic fibrosis in adults. Philadelphia: Lippincott-Raven; 1999:383-418.
32. Floyd-Still JD. Crohn's disease and cystic fibrosis. Dig Dis Sci 1994;39: 880-885.
33. Neglia JP, Fitzsimmons SC, Maisonneuve P, et al. The risk of cancer among patients with cystic fibrosis. Cystic fibrosis and cancer study group. N Engl J Med 1995;332:494-499.
34. Auguiano A, Oates RD, Amos JA, et al. Congenital bilateral absence of the vas deferens: a primarily genital form of cystic fibrosis. JAMA 1992;267: 1794-1797.
35. Chillion M, Casals T, Mercier B, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 1995;331:1475-1480.
36. Mak V, Zielenski J, Tsui LC, et al. Proportion of cystic fibrosis gene mutations not detected by routine testing in men with obstructive azoospermia. JAMA 1999;281:2217-2224.
37. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease; nocturnal oxygen therapy trial group. Ann Intern Med 1980; 93:391-398.
38. Schidlow DV, Taussig LM, Knowles MR. Cystic Fibrosis Foundation consensus conference report on pulmonary complications of cystic fibrosis. Pediatr Pulmonol 1993;15:187-198.
39. Thomas J, Cook DJ, Brocks D. Chest physical therapy of patients with cystic fibrosis: A meta-analysis. Am J Respir Crit Care Med 1995;151:846-850.
40. Elphick HE, Tan A. Single versus combination intravenous antibiotic therapy for people with cystic fibrosis. Cochrane Database Syst Rev 2001; (1):CD002007.
41. Elston C, Geddes D. Inflammation in cystic fibrosis. When and why? Friend or foe? Semin Respir Crit Care Med 2007;28:286-294.
42. Clement A, Tamalet A, Leroux E, et al. Long-term effects of azithromycin in patients with cystic fibrosis: a double-blind, placebo-controlled trial. Thorax 2006;61:895-902.
43. Saiman L. The use of macrolide antibiotics in patients with cystic fibrosis. Curr Opin Pulm Med 2004;10:515-523.
44. Equi A, Balfour-Lynn IM, Bush A, et al. Long-term azithromycin in children with cystic fibrosis: A randomized, placebo-controlled crossover trial. Lancet 2002;360:978-984.
45. Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa. A randomized controlled trial. JAMA 2003;290:1749-1756.
46. Wolter J, Seeney S, Bell S, et al. Effect of long-term treatment with azithromycin on disease parameters in cystic fibrosis: a randomized trial. Thorax 2002;57:212-216.
47. Ramsey BW, Pepe MS, Quan JM, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Engl J Med 1999;340:23-30.
48. Equi A, Balfour-Lynn IM, Bush A, et al. Long-term azithromycin in children with cystic fibrosis. A randomized, placebo-controlled crossover trial. Lancet 2002;360:978-984.
49. Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003;290:1749-1756.
50. Smith A, Cohen M, Ramsey B. Pharmacotherapy. Cystic Fibrosis in Adults. Philadelphia: Lippincott – Raven; 1999:354-364.
51. Suri R. The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs 2005;19:135-144.
52. Desmond KJ, Schwenk WF, Thomas E, et al. Immediate and long-term effects of chest physiotherapy in patients with cystic fibrosis. J Pediatr 1983;103:538-542.
53. Konig P, Gayer D, Barbero GJ, et al. Short-term and long-term effects of albuterol aerosol therapy in cystic fibrosis: a preliminary report. Pediatr Pulmonol 1995;20:205-214.
54. Hordvik NL, Sammut PH, Judy CG, et al. The effects of albuterol on the lung function of hospitalized patients with cystic fibrosis. Am J Respir Crit Care Med 1996;154:156-160.
55. Eggleston PA, Rosenstein BJ, Stackhouse CM, et al. A controlled trial of long-term bronchodilator therapy in cystic fibrosis. Chest 1991;99: 1088-1092.
56. Avital A, Sanchez I, Chernick V. Efficacy of salbutamol and ipratropium bromide decreasing bronchial hyperreactivity in children with cystic fibrosis. Pediatr Pulmonol 1992;13:34-37.
57. Weintraub SJ, Eschenbacher WL. The inhaled bronchodilators ipratropium bromide and metaproterenol in adults with cystic fibrosis. Chest 1989;95:861-864.
58. Sanchez I, Holbrow J, Chernick V. Acute bronchodilator response to a combination of beta-adrenergic and anticholinergic agents in patients with cystic fibrosis. J Pediatr 1992;120:486-488.
59. Elkins MR, Robinson M, Rose BR, et al. for the National Hypertonic Saline in Cystic Fibrosis Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006;354: 229-240.
60. King M, Dasgupta B, Tomkiewicz RP, et al. Rheology of cystic fibrosis sputum after in vitro treatment with hypertonic saline alone and in combination with recombinant human deoxyribonuclease I. Am J Resp Crit Care Med 1997;156:173-177.
61. Wills PJ, Hall RL, Chan W, et al. Sodium chloride increases the ciliary transportability of cystic fibrosis and bronchiectasis sputum in the mucus-depleted bovine trachea. J Clin Invest 1997;99:9-13.
62. Robinson M, Regnis JA, Bailey DL, et al. Effect of hypertonic saline, amiloride and cough on mucociliary clearance in patients with cystic fibrosis. Thorax 1997;52:900-903.
63. Eng PA, Morton J, Douglass JA, et al. Short-term efficacy of ultrasonically nebulized hypertonic saline in cystic fibrosis. Pediatr Pulmonol 1996;21: 77-83.
64. Gilbert DN, Moellering RC, Eliopoulos GM, et al, eds. The Sanford Guide to Antimicrobial Therapy 2007.
65. Konstan MW, Berger M. Current understanding of the inflammatory process in cystic fibrosis: Onset and etiology. Pediatr Pulmonol 1997;24:137-142.
66. Chernick WS, Barbero GJ. Composition of tracheobronchial secretions in cystic fibrosis of the pancreas and bronchiectasis. Pediatr 1959;24:739-745.
67. Potter J, Matthews LW, Lemmon J, et al. Composition of pulmonary secretions from patients with and without cystic fibrosis. Am J Dis Child 1960; 100:493-495.
68. Shak S, Capon DJ, Hellmas R, et al. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci USA 1990;87: 88-92.
69. Armstrong JB, White JC. Liquefaction of viscous purulent exudates by deoxyribonuclease. Lancet 1950;II:739-742.
70. Shak S, Capon DJ, Hellmiss R, et al. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci USA 1990;87: 9188-9192.
71. Wilmott RW, Amin RS, Colin AA, et al. Aerosolized recombinant human DNase in hospitalized cystic fibrosis patients with acute pulmonary exacerbations. Am J Resp Crit Care Med 1996;153:1914-1917.
72. Davies J, Trindale MT, Wallis C, et al. Retrospective review of the effects of rhDNase in children with cystic fibrosis. Pediatr Pulmonol 1997;23: 243-248.
73. Suri R, Grieve R, Normand C. et al. Effects of hypertonic saline, alternate day and daily rhDNase on healthcare use, costs and outcomes in children with cystic fibrosis. Thorax 2002;57:841-846.
74. Rao S, Wilson DB, Brooks RC, et al. Acute effects of nebulization of n-acetylcysteine on pulmonary mechanics and gas exchange. Am Rev Respir Dis 1970;102:17-25.
75. Auerbach HS, Williams M, Kirkpatrick JA, et al. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet 1985;2:686-688.
76. Eigen H, Rosenstein BJ, Fitzsimmons S, et al. A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. Cystic Fibrosis Foundation Prednisone Trial Group. J Pediatr 1995;126:515-523.
77. Pantin CF, Stead RJ, Hodson ME, et al. Prednisolone in the treatment of airflow obstruction in adults with cystic fibrosis. Thorax 1986;41:34-38.
78. Greally P, Hussain MJ, Vergani D, et al. Interleukin-1 alpha soluble interleukin-2 receptor and IgG concentrations in cystic fibrosis treated with prednisone. Arch Dis Child 1994;71:35-39.
79. Eigen H, Rosenstein BJ, Fitzsimmons s, et al. A multicenter study of alternate-day prednisone treatment in children with cystic fibrosis: Cystic Fibrosis Foundation Prednisone Trial Group. J Peditr 1995;126:515-523.
80. Lai HC, FitzSimmons SC, Allen DB, et al. Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N Engl J Med 2000; 342:851-859.
81. Lai HC, FitzSimmons SC, Allen DB, et al. Risk of persistent growth impairment after alternate day prednisone therapy in children with cystic fibrosis. N Engl J Med 2000;342:851-859.
82. Schitotz PO, Jorgensen M, Flensborg EW, et al. Chronic Pseudomonas aeruginosa lung infection in cystic fibrosis: A longitudinal study of immune complex activity and inflammatory response in sputum sol-phase of cystic fibrosis patients with chronic Pseudomonas aeruginosa infections: Influence of local steroid treatment. Acta Paediatr Scan 1983;72:283-287.
83. van Haren EH, Lammers JW, Festen J, et al. The effects of the inhaled corticosteroid budenoside on lung function and bronchial hyperresponsiveness in adult patients with cystic fibrosis. Respir Med 1995;89:209-214.
84. Nikolaizik WH, Schoni MH. Pilot study to assess the effect of inhaled corticosteroids on lung function in patients with cystic fibrosis. J Pediatr 1996; 128:271-274.
85. Davis PB. di Sant'Agnese PA. Assisted ventilation for patients with cystic fibrosis. JAMA 1978;239:1851-1854.
86. Sood N, Paradowski LJ, Yankaskas JR. Outcomes of intensive care unit in adults with cystic fibrosis. Am J Respir Crit Care Med 2001;163:335-338.
87. Efrati O, Kremer MR, Barak A, et al. Improved survival following lung transplantation with long-term use of bilevel positive pressure ventilation in cystic fibrosis. Lung 2007;183:73-79.
88. Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary fluid liquid layer depletion, not normal ion composition in the pathogenesis of cystic fibrosis airways disease. Nocturnal nasal intermittent positive pressure ventilation (BiPAP) in respiratory failure. Chest 1992;101:516-521.
89. Waldhorn RE. Nocturnal nasal intermittent positive pressure ventilation with bi-level positive airway pressure (BiPAP) in respiratory failure. Chest 1992; 101:516-521.
90. Efrati O, Modan-Moses D, Barak A, et al. Long-term non-invasive positive pressure ventilation among cystic fibrosis patients awaiting lung transplantation. Isr Med Assoc J 2004;6:527-530.
91. Rodrigo G, Pollack C, Rodrigo C, et al. Heliox for nonintubated acute asthma patients. Cochrane Database Syst Rev 2006; (4):CD002884.
92. Henchey K. Use of heliox therapy to relieve acute respiratory distress in an adolescent with cystic fibrosis: A case report. AM J Crit Care 2003;12: 556-557.
93. Tirouvanziam R, Conrad CK, Bottiglieri T, et al. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc Natl Acad Sci 2006;103:4628-4633.
94. Robinson W. Palliative care in cystic fibrosis. J Palliat Med 2000;3:187-192.
With advances in medical science, patients with serious congenital diseases are living into adulthood. Where once cystic fibrosis patients died in infancy, patients now live into their 40s. Although many of the standard treatments for COPD apply to cystic fibrosis, there are specific differences in management, which this monograph highlights.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.