Inborn Errors of Metabolism in Infants: Recognition, Diagnosis, and Management
Inborn Errors of Metabolism in Infants: Recognition, Diagnosis, and Management
Authors: Glenn D. Harris, MD, Associate Professor of Pediatrics; Director, Pediatric Diabetology, Brody School of Medicine at East Carolina University, Greenville, NC; and Ronald M. Perkin, MD, MA, FAAP, FCCM, Professor and Chairman, Department of Pediatrics, Brody School of Medicine at East Carolina University; Medical Director, University Health Systems of Eastern Carolina Children’s Hospital, Greenville, NC.
Peer Reviewer: Alfred Sacchetti, MD, FACEP, Director of Research, Department of Emergency Medicine, Our Lady of Lourdes Hospital, Camden, NJ.
Inborn errors of metabolism (IEM) often present as acute episodes that require hospital attention and emergency care. The clinical suspicion of an IEM is essential, since the collection of appropriate laboratory samples during an episode of clinical decompensation is the key to a specific diagnosis. However, the early recognition of IEMs is difficult, probably because they occur infrequently, and their predominant signs and symptoms are nonspecific.
In the emergency department (ED), clinicians faced with an acutely ill child routinely do not suspect an IEM until the more common conditions have been excluded.1-16 Consequently, diagnoses and specific therapeutic measures are delayed or missed; frequently, the appropriate samples needed for the specific diagnoses are not collected. Another important factor is that many studies have focused their objectives on the description of the clinical phenotypes, not the acute presentation of the disease or common features of the first decompensation.6
Patients often arrive to the ED with symptoms requiring immediate attention, such as cardiorespiratory arrest, apnea, seizures, or coma. Studies have shown a predominance of neurologic signs followed by digestive symptoms. Fewer than 10% of patients with IEM present without neurologic or digestive signs.1,2 Lethargy, coma, seizures, and abnormalities in muscle tone are the common neurologic presentations, while vomiting, feeding difficulties, and hepatic dysfunction are the predominant digestive signs and symptoms.
It is remarkable that most patients with severe metabolic decompensation, especially when complicated by apnea and cardiorespiratory arrest, are young (< 2 years of age).1
Many inborn errors of intermediary metabolism present during periods of poor oral intake or intercurrent illness.3,17 Under these conditions, the body switches to a predominantly catabolic state, in which glycogen, fat, and protein are mobilized as alternative energy sources. If a genetic defect already exists in any of these catabolic pathways, the problem is accentuated, because the catabolic state demands a high flux of metabolite through these pathways. Therefore, a cardinal principle in the management of children who have IEM is to prevent them from lapsing into a catabolic state.
The number, complexity, and varied clinical presentations of IEM present a formidable challenge to the practicing physician. Yet, in many cases, prevention of death or permanent neurologic sequelae in patients with these disorders is dependent on early diagnosis and institution of appropriate therapy. It is, therefore, incumbent on the ED physician to be familiar with the major signs and symptoms of IEM and with the basic laboratory studies necessary to arrive at an initial diagnosis. Waiting until sepsis and other, more common causes of illness are ruled out before initiating a specific diagnostic evaluation is inadvisable, as is indiscriminate study of all ill newborns for metabolic disorders.
It is the purpose of this article to define the constellation of findings in the young infant that should alert the physician to the possibility of inherited metabolic disease. The discussion is confined to those disorders that typically present in early infancy and have potential life-threatening consequences. The treatment of important groups of metabolic disorders is addressed, focusing on stabilization and acute management of patients with these conditions.It is hoped that a presentation of the essential biochemical pathways will facilitate understanding of the correlation between biochemical derangements and the clinical presentations of a variety of IEM.— The Editor
General Concepts in Energy Metabolism
Energy in the form of glucose supplies the majority of the body’s needs under normal homeostasis. At the cellular/molecular level, the end point of glucose metabolism is generation of adenosine triphosphate (ATP). When the supply of glucose cannot be maintained by dietary intake or by utilization of glycogen stores, a catabolic state ensues, with utilization of fat and protein to furnish the necessary ATP. Fat and protein catabolism, as well as glucose metabolism induce generation of ATP via the Krebs or tricarboxylic acid cycles which convert acetyl coenzyme A (acetyl CoA) to the reduced form of nicotinamide adenine dinucleotide (NADH). NADH undergoes oxidative phosphorylation within the mitochondria, where the chemical energy of NADH is transferred to ATP.3 If catabolism is not arrested by appropriate treatment, starvation ensues. If adequate energy cannot be produced, death occurs. Patients with severe forms of some IEMs may die even with appropriate recognition and treatment.
Glucose and Other Carbohydrates. Through the process called glycolysis, glucose is converted to pyruvate, a 3-carbon ketoacid that is an important intermediary substance. It has many different fates that depend on the metabolic status of the cell. It can be converted to and exists in equilibrium with lactate; it can be used to make glucose through a process called gluconeogenesis; and it is converted to acetyl CoA, a 2-carbon compound that then is fed into the Krebs cycle.
The most important function of the Krebs cycle is to convert acetyl CoA to NADH. Captured within NADH is the major portion of the chemical energy from glucose. NADH then is carried through a cascade of reactions within the mitochondria, known as oxidative phosphorylation, in which the chemical energy locked within NADH is transferred to ATP. Thus, through the successive steps of glycolysis, Krebs cycle, and oxidative phosphorylation, glucose is used to make ATP.3
It is interesting to note that the equilibrium between pyruvate and lactate mentioned previously depends on the availability of NADH. When NADH is in excess, lactate is favored; when NADH is in short supply, pyruvate is favored, such that more pyruvate is available to generate NADH.
Because the daily diet rarely consists of pure glucose as the carbohydrate source, many metabolic pathways are needed to convert other carbohydrates to glucose. Therefore, carbohydrates such as fructose, galactose, lactose, sucrose, and starch eventually are converted to glucose if they are to be used to generate energy. These conversion processes are determined genetically. Failure of a conversion process because of a genetic defect often leads to accumulation of intermediates, some of which may be toxic to the body.
Another problem with the inability to convert other carbohydrate sources to glucose would appear to be the deprivation of a potential energy source to the body. This problem is most dramatic when the defect involves the inability to convert endogenous carbohydrate — glycogen — to glucose.
Fat, Acetyl CoA, and Ketones. Of all the compounds that represent energy reserve for the body, fat has the highest caloric content per molecule. Therefore, it is not surprising that mobilization of fat is one of the key adaptive biochemical responses when glucose and glycogen sources are exhausted for ATP generation. Fat is composed of a 3-carbon backbone (a glycerol derivative) coupled to long-chain fatty acids with varying degrees of saturation. The majority of the chemical energy is stored in the long-chain fatty acids. During fat breakdown, the glycerol and the long-chain fatty acids are separated first. Metabolism of the long-chain fatty acids requires the successive removal of 2-carbon units from the carboxyl end of the molecules through a process called beta-oxidation.3
As nature would have it, the 2-carbon unit that is removed from the fatty acid is acetyl CoA, a key intermediate in the pathway from glucose to ATP. Therefore, the metabolism of fatty acid dovetails into the existing pathway of energy metabolism, with the generated acetyl CoA being fed into the Krebs cycle for eventual NADH and ATP production. Another noteworthy biochemical feature is that, during fat breakdown, the massive flux of acetyl CoA results in its higher steady-state level. Because ketone bodies can be thought of as dimers of acetyl CoA, a consequence of having higher levels of acetyl CoA is a higher rate of ketone production. Therefore, ketosis is a biochemical hallmark of effective fat mobilization.
Protein, Amino Acids, Organic Acids, and Ammonia. The mobilization of fat during the catabolic state almost always is accompanied by mobilization of protein as an energy source. Protein is made up of amino acids, which can be thought of as being composed of two components: the amino group and an organic acid backbone. The amino group is metabolized in the form of ammonia, which can accumulate to toxic levels if not disposed of efficiently. This is done through the urea cycle, which takes place in the liver. The net effect of the urea cycle is conversion of excess ammonia into readily excretable urea.
The organic acid backbone is more complicated. The many different amino acids have many different configurations of the organic acid backbone, and each organic acid backbone is metabolized somewhat differently. Fortunately, a thread of logic governs these diverse catabolic pathways: All of the end products of the organic acid metabolism pathways are compounds that fit into the existing scheme of energy metabolism. Many of these organic acids yield acetyl CoA or ketone bodies during their catabolism, and some are converted to pyruvate. Thus, during protein mobilization, the chemical energy stored in amino acids is harnessed in the generation of ATP through the Krebs cycle and oxidative phosphorylation.3
Another feature of amino acid metabolism that is not involved directly in energy metabolism is the conversion of some amino acids into other amino acids. When this conversion fails because of a genetic defect, the substrate amino acid accumulates to toxic levels. This is exemplified by phenylketonuria, in which phenylalanine fails to convert to tyrosine. A high blood phenylalanine level ensues, which is toxic to the central nervous system and leads to mental retardation.
IEM: General Overview
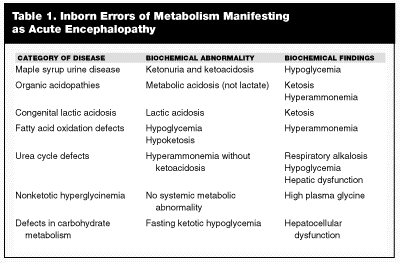
IEM are a heterogeneous group of disorders in which a single gene deficit results in an enzyme or transport protein deficiency, causing a block in a metabolic pathway.4 The clinical abnormalities occur from either accumulation of the metabolic substrate behind the block and/or deficiency of an end product. Both males and females are affected since the gene responsible for an autosomal recessive disorder is located on one of the 22 autosomes.8 The clinical manifestations or phenotype of the disorders are determined by the severity of the particular genetic abnormality and the associated environmental factors, such as diet or intercurrent infection.17 More than 400 IEMs have been identified, a minority of which will present in the infant or young child as an acute, serious, or life-threatening event.6 Inborn errors of metabolism have been categorized in various ways, with no classification being universally accepted because the disease categories often overlap. Classifications can be based on categories of underlying defect and/or by initial biochemical presentation. (See Figure 1.)
IEM are rare, nearly always autosomal recessive or, less often, X-linked recessive disorders.6 However, if taken as a group of disorders, they are not rare; cumulatively, IEM may occur in more than 1 in 5000 live births.8,10 It is incumbent on the clinician to recognize the signs and symptoms of these disorders so that early, appropriate laboratory testing and treatment are provided prior to the advent of a life-threatening event. In addition, obtaining a family history for IEM, consanguinity, unexplained childhood deaths, or acute life-threatening events may be paramount to the clinician to facilitate early diagnosis of an IEM. Missing the diagnosis may result in otherwise preventable morbidity, life-long disability, or death. Some disorders may have relatively simple treatments such as avoiding certain foods or adding vitamin supplements; for example, biotin supplementation for biotinidase deficiency may be all that is required for maintaining health.12
Newborn Screening. Approximately 4 million infants are born yearly in the United States and are screened to detect conditions that threaten their lives and long-term health.18,19 Newborn screening is a public health activity aimed at the early identification of infants who are affected by certain genetic, metabolic, or infectious conditions. Early identification of these conditions is particularly crucial, as timely intervention can lead to a significant reduction of morbidity, mortality, and associated disabilities in affected infants.
In all states today, every newborn is screened for two disorders: phenylketonuria (PKU) and hypothyroidism.18 Beyond these two tests, there is inconsistency between states in the panel of conditions screened. Many states screen for various additional disorders such as maple syrup urine disease, homocystinuria, tyrosinemia, galactosemia, biotinidase deficiency, medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, and sickle cell anemia. When an infant presents with a severe illness or life-threatening event, the newborn screen should be reviewed.
Clinical Manifestations of IEM
Acute Metabolic Encephalopathy. Several groups of inherited metabolic disorders, most notably the organic acidemias, urea cycle defects, and certain disorders of amino acid metabolism, typically present with acute life-threatening symptoms of an encephalopathy.2,7 (See Table 1.) These symptoms are the result of toxic effects of accumulating metabolites on the central nervous system (CNS). Because most of these metabolites cross the placenta and are cleared by the mother during gestation, affected infants usually appear normal at birth. The interval between birth and onset of clinical symptoms ranges from hours to months. The initial findings usually are those of lethargy and poor feeding, as seen in almost any sick infant. Although sepsis often is the first consideration in infants who present in this way, these symptoms in a full-term infant with no specific risk factors for sepsis strongly suggest a metabolic disorder. Infants with inborn errors of metabolism may become debilitated and septic rather quickly, and it is therefore important that the presence of sepsis not exclude consideration of other possibilities. If untreated, the lethargy associated with these conditions may progress to coma. Other signs of CNS dysfunction, such as seizures and abnormal muscle tone, also may be noted. Evidence of cerebral edema may be observed, and intracranial hemorrhage occasionally occurs.2,7
 |
Subdural and retinal hemorrhages in young children without an appropriate history of trauma strongly suggest non-accidental injury. Similar features are occasionally found in patients with glutaric aciduria type 1 (GA1), a rare inborn error of metabolism, and have led to the misdiagnosis of non-accidental injury.20,21
GA1 is an autosomal recessive disorder caused by deficiency of the enzyme glutaryl-CoA dehydrogenase. It commonly presents before age 18 months with sudden onset of encephalopathy, followed by a severe and persistent movement disorder.20,22 Clinical examination at this stage often shows macrocephaly, and cerebral imaging may show bilateral frontotemporal atrophy or widening of the Sylvian fissure, with or without subdural effusions.20,22 Subdural hematomas have been found in symptomatic and asymptomatic patients.20 They can occur with minimal trauma presumably because the bridging veins are elongated in the presence of cerebral atrophy and easily ruptured.20 Retinal hemorrhages also have been reported.20
An infant with an IEM may present with the gradual development of lethargy and poor feeding or abruptly with apnea or respiratory distress.2 The apnea is typically central in origin and a symptom of the metabolic encephalopathy, but tachypnea may be a symptom of an underlying metabolic acidosis, as occurs with the organic acidemias. Infants with urea cycle defects and evolving hyperammonemic coma initially exhibit central hyperventilation, which leads to respiratory alkalosis. Ammonia is a potent stimulant of the respiratory center, and hyperammonemia results in varying degrees of respiratory alkalosis.3,14,23
IEM appear in newborns and infants in one of two forms — acute, fulminant or chronic, slowly progressive disorders. The most common acute encephalopathies fall into one of seven categories. These include maple syrup urine disease, organic acidopathies, congenital lactic acidosis, fatty acid oxidation defects, urea cycle disorders, nonketotic hyperglycinemia, and disorders of carbohydrate metabolism. (See Table 1.) Chronic progressive disorders are much less common and fall into one of the following disease groups: peroxisomal disorders, pyridoxine dependency, hyperphenylalaninemias, and some forms of lysosomal storage diseases.
The first step in identifying metabolic encephalopathies is to think of them. Metabolic disease should be considered in the differential diagnosis of any newborn or infant demonstrating symptoms or signs of acute or progressive encephalopathy. An acute or progressive encephalopathy is characterized by any combination of symptoms and signs of diffuse central nervous system dysfunction that evolve in severity over hours or days. What is characteristic of metabolic disease is that the overall pattern and temporal evolution are generally dominated by progressive depression of consciousness, and motor abnormalities and brainstem dysfunction parallel the severity of disturbed consciousness in a manner that does not localize to a particular neuroanatomic structure.14
Suspicion of an IEM should be elevated by the presence of a variety of nonneurologic abnormalities. Prominent, unexplained vomiting, hyperpnea in the absence of lung disease, unusual odors, or other organ involvement (i.e., cataracts; sparse, brittle hair; unusual skin rashes such as ichthyosis; hepatospleno-megaly; liver failure; cardiomyopathy; neutropenia; or thrombocytopenia) may appear in the course of decompensation of an IEM.6 A careful family history should be obtained to ascertain consanguinity, known inherited metabolic disease, excess fetal loss, or infant demise in a sibling under similar circumstances.
Once an infant is identified as having an acute encephalopathy, further evaluation should proceed urgently to determine its cause. The most common causes usually can be suspected based on history, known risk factors, and existing diagnoses. These include hypoxic-ischemic injury, intracranial hemorrhage, central nervous system infection, and intoxication. Diagnosis is further delineated by appropriate first-stage laboratory studies as shown in Table 2.13 The aim of the initial evaluation is to simultaneously narrow the diagnosis and treat potentially life-threatening or handicapping conditions as quickly as possible. Results of first-stage studies should reveal abnormalities requiring immediate corrections, such as hypoglycemia, hyponatremia, hypocalcemia, or hypoxia. The studies also should further delineate other organ involvement, such as renal or hepatocellular dysfunction. A constellation of abnormalities on the initial screen may suggest a particular IEM. (See Figure 1 and Figure 2.)
 |
For example, severe metabolic acidosis combined with hyperammonemia and ketotic hypoglycemia would strongly suggest an organic acidemia, such as propionic acidemia, whereas a respiratory alkalosis combined with severe hyperammonemia without ketotic hypoglycemia would likely be a urea cycle disorder. If the results of first-stage assessment combined with clinical history, examination, and neuroimaging do not provide a specific cause for encephalopathy, they should provide the rationale for choosing appropriate second-stage studies. These studies are aimed at identifying the category of metabolic defect and in some cases provide a probable specific diagnosis that can be verified in the third stage, in which tissue sampling provides material for enzyme quantification or genetic analysis.
Hypoglycemia. Hypoglycemic encephalopathy is rarely an isolated or pure insult in newborns and infants. However, hypoglycemia is a common finding in many critically ill infants and is a frequent manifestation of IEM.
The brain is a principal consumer of circulating glucose and, for all practical purposes, is dependent on glucose alone for its metabolic needs. When the brain is deprived of glucose for prolonged periods or on multiple occasions, injury occurs.24,25
The neurologic signs and symptoms attributable solely to deficient supply to the brain of glucose of glucose-sparing substrates have been defined.26 The early stages may be dominated by signs of elevated circulating catecholamines: tachycardia, pallor, diaphoresis, irritability, jittery movements or tremors, and exaggerated tendon reflexes and neonatal reflexes (Moro reflex). As cerebral glucose stores decline, there ensues progressive depression of consciousness, hypotonia, depressed reflexes, and often seizures. At its worst, uncorrected hypoglycemia in the absence of circulating ketone bodies will progress to coma, characterized by an isoelectric electroencephalogram, cerebral edema, flaccid unresponsiveness, apnea, and loss of brainstem reflexes.26 Hypothermia and myocardial dysfunction may accompany the intermediate and severe stages. If not treated, hypoglycemic coma is associated with irreversible neuronal necrosis and lasting neurologic deficits, the severity of which depend on the duration of coma.
A number of factors make the infant and child more vulnerable to hypoglycemia than the adult.26,27 Glucose utilization is higher in the child, probably due to the proportionately larger brain.24 Glycogen stores are smaller and may only be sufficient to provide circulating glucose for a fast of a few hours. Available muscle mass and fat stores from which to generate precursors for gluconeogenesis and ketogenesis are smaller.
Any disorder that interferes with carbohydrate intake, intestinal absorption, glycogen formation or mobilization, gluconeogenesis, ketogenesis, or fatty acid oxidation places a child at risk for hypoglycemia.24 Similarly, disorders that result in excessive tissue utilization of glucose (e.g., hyperinsulinism) place a child at risk for hypoglycemia.
Hypoglycemia and its associated symptoms occasionally may be seen in infants with disorders of protein intolerance, but it is seen more commonly in disorders of carbohydrate metabolism or fatty acid oxidation.2 Among the best known IEMs associated with hypoglycemia are the hepatic glycogen storage diseases (GSD). The hypoglycemia in these disorders is related to the inability of the liver to release glucose from glycogen, and it is most profound during periods of fasting. Hypoglycemia, hepatomegaly, and lactic acidosis are prominent features of these disorders. Hypoglycemia is not a feature of GSD type II (Pompe disease) because cytoplasmic glycogen metabolism and release are normal in this disorder in which glycogen accumulates within lysosomes as a result of deficiency of the enzyme acid maltase. Clinical manifestations of this disorder include macroglossia, hypotonia, cardiomegaly with congestive heart failure, and hepatomegaly. Cardiomegaly is the most striking feature and may be apparent in the neonatal period.28 Congestive heart failure is the cause of death in most cases. Hypoglycemia may be a prominent feature of both galactosemia and hereditary fructose intolerance, although symptoms of the latter disorder occur only after fructose (sucrose) has been introduced to the diet.
A number of inherited defects in fatty acid oxidation have been identified in infants presenting with hypoglycemia.29-31 These disorders are important because of their apparent frequency and because of the variability of the initial presentation. Infants affected have an impaired capacity to use stored fat for fuel during periods of fasting and readily deplete their glycogen stores. Despite the development of hypoglycemia, acetyl-CoA production is diminished, and ketone production is impaired. The hypoglycemia occurring in these conditions is typically characterized as nonketotic, although small amounts of ketones may be produced.29 Hypoglycemia may occur as an isolated finding or may be accompanied by many of the other biochemical derangements typically associated with Reye syndrome, such as hyperammonemia, metabolic acidosis, and elevated transaminases.32 Hepatomegaly may or may not be present. Any infant presenting with findings suggesting Reye syndrome should be evaluated for fatty acid oxidation defects.
The most common of the fatty acid oxidation defects is MCAD deficiency. In addition to presenting as nonketotic hypoglycemia or a Reye’s-like syndrome, it may present as sudden death or an acute life-threatening event.15,16,33 Many reports of infants diagnosed as having MCAD deficiency have described a history of a sibling who died of SIDS. Fat accumulation in the liver or muscle of any infant dying unexpectedly should strongly suggest the possibility of this or a related disorder of fatty acid oxidation.29 Very long-chain fatty acyl-CoA dehydrogenase deficiency is associated with similar clinical findings, although there also may be evidence of a cardiomyopathy. Infants with this and several other fatty acid oxidation defects may present with cardiac arrhythmias or unexplained cardiac arrest.31
The accumulation of fatty acyl-CoAs in patients with fatty acid oxidation defects leads to a secondary carnitine deficiency, probably as a result of excretion of excess acylcarnitines in the urine. Urine organic acid analysis, measurement of serum carnitine, and analysis of the plasma acylcarnitine profile are the most helpful laboratory studies in the initial screening for defects in fatty acid oxidation.30 These studies are sufficient to establish the diagnosis of MCAD deficiency, which is associated with the presence of a characteristic metabolite, octanoylcarnitine, on the acylcarnitine profile.30 Enzymatic assays may be necessary for the definitive diagnosis of some of the fatty acid oxidation defects. As is true for the defects in carbohydrate metabolism leading to hypoglycemia, treatment of the fatty acid oxidation defects involves avoidance of fasting and provision of adequate glucose. Restriction of dietary fat intake and supplemental L-carnitine therapy are recommended.13,17
Glutaric acidemia type II is an IEM involving fatty acid and amino acid oxidation.33 Affected infants present clinically with hypoketotic hypoglycemia, severe metabolic acidosis, and a characteristic organic aciduria. There are two forms of the disease: severe neonatal and mild late-onset. In the more severe form, patients present during the neonatal period and die within the first month of life. Patients with the relatively mild, late-onset form of the disease present later in life (months to years) and may only exhibit symptoms and diagnostic metabolite abnormalities intermittently during periods of stressed catabolic states caused by fasting or intercurrent illness.
When any infant develops signs and symptoms of acute encephalopathy, a complete first-stage evaluation should be performed. Blood glucose values of less than 45 mg/dL (2.5 mmol/L) should be treated while the evaluation proceeds. If ketones are absent and the exogenous glucose requirement is high (greater than 10 mg/kg/min), then hyperinsulinism should be suspected and further evaluated by measuring insulin levels.24 This is a logical means to quickly narrow down the differential diagnosis by means of simple bedside findings and first-stage laboratory studies that determine the presence or absence of the following findings: 1) hepatomegaly; 2) ketonuria; 3) metabolic acidosis; and 4) lactic acidemia.26 It is important that key laboratory studies, such as urine for ketones, plasma lactate, and acid-base status, be obtained at the same time that hypoglycemia and its symptoms are occurring. Additional information from the clinical setting further suggests the likelihood of certain diseases, such as the presence of myopathic weakness and cardiomyopathy in the case of glycogen storage disease, or the appearance of hyponatremic, hyperkalemic, or hypovolemic shock in the case of adrenal insufficiency.
Treatment of acute, symptomatic hypoglycemia should proceed immediately after its identification. Recent recommendations have shifted away from the use of hyperosmolar dextrose solutions in favor of a minibolus of 250 mg/kg using 10% dextrose infused over 2 minutes.26,27 This minibolus should be followed by continuous infusion of 10% dextrose starting at 8 mg/kg/min and increasing as necessary to maintain stable blood-glucose levels of greater than 45 mg/dL. Monitoring the effect of glucose administration on the clinical symptoms, particularly level of consciousness, can help clarify the neurologic significance of the hypoglycemia and the need for evaluation for other causes of encephalopathy. Discontinuation of intravenous glucose infusion should proceed gradually, with frequent blood-glucose monitoring to avoid the recurrence of hypoglycemia.
Hyperammonemia. Among the most important findings associated with IEM presenting with an acute encephalopathy is hyperammonemia.2,23 A plasma ammonia level should be obtained for any child with unexplained vomiting, lethargy, or other evidence of an encephalopathy.23,34-36 Significant hyperammonemia is observed in a limited number of conditions, including urea cycle defects and many of the organic acidemias. Also in the differential diagnosis in the neonate is a condition referred to as transient hyperammonemia of the newborn (THAN), whereas in the older infant, fatty acid oxidation defects may be considered.2 Ammonia levels in newborns with these conditions frequently exceed 1000 mcg/mol/L.35 The finding of marked hyperammonemia provides an important clue to diagnosis and indicates the need for urgent treatment to reduce the ammonia level. The degree of neurologic impairment and developmental delay observed subsequently in affected infants has been shown to be dependent on the duration of the neonatal hyperammonemic coma.14,34
The timing of the onset of symptoms may provide an important clue. Patients with some of the organic acidemias, such as glutaric acidemia type II or pyruvate carboxylase (PC) deficiency, may exhibit symptomatic hyperammonemia during the first 24 hours of life. Symptoms in the first 24 hours also are characteristic of THAN, a condition that is poorly understood but, apparently, not genetically determined.
Infants who develop severe hyperammonemia after 24 hours of age usually have a urea cycle defect or an organic acidemia; infants with organic acidemias typically exhibit a metabolic acidosis as well. Urine organic acid analysis always should be obtained, regardless of whether acidosis is present. Because urea cycle intermediates are amino acids that generally are basic in nature, anion gap metabolic acidosis is not seen unless there is a significant degree of dehydration secondary to the acute illness. Plasma amino acid analysis is helpful in the differentiation of the specific defects in this group. In addition, carbamyl phosphate synthetase deficiency and ornithine transcarbamylase (OTC) deficiency may be differentiated by measuring urine orotic acid, which is low in the former and elevated in the latter.23,36 Although the family history often is negative, a positive history of early male deaths or of females with episodic illness in the family of a male infant with hyperammonemia suggests ornithine transcarbamylase deficiency, the only one of the urea cycle defects with a sex-linked mode of inheritance.36
Urea cycle disorders are characterized by the triad of encepha-lopathy, respiratory alkalosis, and hyperammonemia.23 As a group, urea cycle disorders may be as common as many well-known diseases.23 It is apparent that OTC deficiency is the most common of the urea cycle disorders, presumably because it is inherited as an X-linked trait (the others are inherited as autosomal recessive traits) and is expressed in some heterozygous females.36
Less significant elevations of plasma ammonia than those associated with IEM and THAN can be observed in a variety of other conditions associated with liver dysfunction, including sepsis, generalized herpes simplex infection, and perinatal asphyxia.2 Liver function studies should be obtained for evaluating the significance of moderate elevations of plasma ammonia. However, even in cases of severe hepatic necrosis, it is rare for ammonia levels to exceed 500 mcg/mol/L.
Hyperammonemia also may result from urea cycle impairment following hepatic failure, malignant disease, or toxin exposure.37 A greatly increased urea load, such as that produced by Proteus and other urea-splitting bacteria, may occur as a result of increased production and absorption of ammonia as NH3 from a reservoir of infected stagnant urine with an increased pH. Hyperammonemia may complicate infectious processes with purulent collections in the peritoneal cavity.
Both acute and chronic hyperammonemias have deleterious effects on CNS function.38 Neurological symptoms range from mild personality disorders to seizures, coma, and mental retardation depending upon the age of the patient and the severity and duration of the hyperammonemia. Neuropathological damage similarly ranges from subtle astrocytic changes to cerebral atrophy and delayed myelination.
Many mechanisms have been proposed to explain the toxic effects of hyperammonemia on brain development and function.38
Precisely which mechanism is operative in a given pathology will depend not only on the degree and duration of hyperammonemia but also on the brain/blood ammonia concentration ratio, a parameter that may be modified by such factors as blood-brain barrier permeability and pH changes.38
Aggressive therapy of hyperammonemia is indicated with the aim of reducing the concentration of blood ammonia as quickly as possible. In patients with severe elevations of ammonia, the preferred method of clearance is hemodialysis.35,36
Metabolic Acidosis. Another important laboratory feature of many of the inborn errors of metabolism during acute episodes of illness is metabolic acidosis with an increased anion gap, readily demonstrable by measurement of arterial blood gases or serum electrolytes and bicarbonate.39 An increased anion gap (> 16) is observed in many IEM and in most other conditions producing metabolic acidosis in the neonate.12 In contrast, the differential diagnosis of metabolic acidosis with a normal anion gap is essentially limited to two conditions, diarrhea and renal tubular acidosis. Among the inborn errors, the largest group typically associated with overwhelming metabolic acidosis in infancy is the group of organic acidemias, including such entities as methylmalonic acidemia, propionic acidemia, and isovaleric acidemia.40
Organic acidemias are a biochemically heterogenous group of IEM that are characterized by the accumulation in tissues and body fluids of amino acid, fatty acid, and carbohydrate metabolism intermediates, conventionally referred to as organic acids.5 If one considers the dicarboxylic acidemias and primary lactic acidemias as being separate entities, the majority of the organic acidemias known involve the metabolism of the essential branch-chain amino acids isoleucine, leucine, and valine.
A combination of metabolic acidosis, elevated anion gap, and rapidly progressing neurological symptoms always should be considered a strong diagnostic indicator of one of these diseases. The presence of ketonuria provides an additional clue toward the recognition of the organic acidemias. Especially in the neonatal period, the presence of significant ketonuria always should be taken as a specific indicator of one of these relatively frequent disorders.5,12 Ketonuria is rare in the normal or severely ill newborn, even when the blood sugar is severely depressed. Urine ketones in the newborn are virtually diagnostic of an IEM such as maple syrup urine disease or methylmalonic or propionic acidemias.
In addition to specific organic acid intermediates, plasma lactate often is elevated in organic acidemias as a result of secondary interference with co-enzyme A (CoA) metabolism.5 Neutropenia and thrombocytopenia are commonly observed and further underscore the clinical similarity of these disorders to neonatal sepsis.5 Hyperammonemia, hypoglycemia, and hyperuricemia are additional informative findings, especially during episodes of decompensation.
Methylmalonic (MMA) and propionic (PA) acidemias are the most common clinically relevant inborn errors of organic acid metabolism in man, with incidences of about 1/50,000 and 1/100,000 live births, respectively.40 Both typically present with ketoacidosis in the neonatal period after a normal delivery and a normal first 1-2 days of life. Some patients are diagnosed later in the first year with failure to thrive, typically with recurrent episodes of vomiting and acidosis. Of clinical interest is the ever-increasing range of complications of these conditions. Basal ganglia infarction is now well-recognized and causes a choreoathetoid movement disorder. Presentation with this feature has been described even in adulthood, and urinary organic acid analysis in children with unexplained choreoathetosis should be routine. Recurrent pancreatitis and renal tubular and glomerular dysfunction both appear to be relatively frequent complications of MMA in particular, but thus far these have only been described in children in whom the organic acidopathy previously has been well-documented. Cardiomyopathy also has been mentioned as a possible association with MMA. Many of the clinical features and complications appear attributable to the recognized ability of propionate to inhibit mitochondrial function; indeed, the alterations in acid-base balance result from lactate and ketone accumulation secondary to inhibition of mitochondrial energy cycles, rather than through the direct effect of increased propionic (or methylmalonic) acid concentration per se.40
Maple syrup urine disease (MSUD), also called branched chain ketoaciduria, is a disorder of amino acid metabolism.5 The enzyme deficient in MSUD, the branched-chain alpha-ketoacid dehydrogenase complex, results in the accumulation of the ketoacids derived from the transamination of the three branched chain amino acids, leucine, isoleucine, and valine. The accumulation of the branched chain amino acids and the ketoacids derived from them exert neurotoxic effects.26 Clinical symptoms and signs in the classic MSUD often manifest toward the end of the first week of life and consist of somnolence, vomiting, seizures, and increased muscle tone. A maple syrup aroma emanates from the urine by the second or third week of life. Biochemical abnormalities include ketoacidosis, ketonuria and, in some cases, hypoglycemia. Diagnosis is accomplished by newborn screening in a few states.18,19 More commonly, identification of the ketoacids in the urine or elevation of isoleucine, valine, and leucine in the blood establishes the presumptive diagnosis.
Defects in pyruvate metabolism or in the respiratory chain may lead to primary lactic acidosis presenting as severe metabolic acidosis in infancy.41-44 Unlike most of the other conditions presenting acutely in the newborn, clinical features of these disorders are unrelated to protein intake. Disorders in this group should be considered in patients with lactic acidosis who have normal urine organic acids. Differentiation of the various disorders in this group can be facilitated by measuring plasma pyruvate and calculating the lactate/pyruvate ratio.2,5 A normal ratio (< 25) suggests a defect in pyruvate dehydrogenase (PDH) or in gluconeogenesis, and an elevated ratio (> 25) suggests PC deficiency, a respiratory chain defect, or a mitochondrial myopathy.
Provided that the oxygen supply is adequate, the bulk of energy requirements for most cell types is generated in mitochondria by oxidation of pyruvate, fatty acids, or ketone bodies. These substances enter as acetyl-CoA into the citric acid cycle and the respiratory chain. Impairment of oxidative metabolism may occur due to disorders of substrate transport, substrate utilization, citric acid cycle function, or the respiratory chain.41 Tissues that are most reliant on oxidative metabolism, including brain skeletal muscle and cardiac muscle, are most jeopardized by disorders of oxidative metabolism, which produce a variety of symptoms and signs related to their dysfunction. Lactic acidemia, with or without metabolic acidosis, is a frequent biochemical marker for disorders of oxidative metabolism, as are hypoglycemia and hyperammonemia.
In the first six months of life, disorders of mitochondrial metabolism fall into three main groups: defects of fatty acid oxidation, defects of pyruvate metabolism, and defects of the respiratory chain, all of which can produce severe brain dysfunction.44
Seizures. Many metabolic perturbations can provoke seizures. Metabolic derangements (i.e., hypoglycemia, hypoxemia, and electrolyte disturbances), systemic diseases (i.e., uremia, liver failure, and hypertension), or IEM (i.e., pyridoxine dependency, MSUD, hyperglycinemia, urea cycle defects, and other amino and organic acid disorders) all may result in seizures. These disturbances transiently alter the microenvironment of the neuron such that excitation is facilitated or inhibition is impaired. Normally, correction of the underlying disorder or removal of the toxin corrects the problem. The risk of subsequent unprovoked seizures is low unless there is a secondary complication that alters the structure of the brain.
Ordinarily, seizures are not the only manifestation of inborn errors of metabolism.45 Seizures usually occur within the context of acute encephalopathy with acidosis, hypoglycemia, hyperammonemia, and other derangements. The exception is pyridoxine dependency, in which seizures are the primary neurologic complication.45
Glycine encephalopathy should be suspected in any infant who has intractable seizures, especially with hiccoughs.4
Vomiting. Vomiting is a striking feature of many of the inborn errors of metabolism associated with protein intolerance, although considerably less common in the newborn than in the older infant. If persistent vomiting occurs in the neonatal period, it usually signals significant underlying disease. Inborn errors of metabolism should always be considered in the differential diagnosis. Formula intolerance is frequently suspected, and many affected infants have numerous formula changes before a diagnosis is finally established.
In the organic acidemias, the accumulating toxic compounds appear to cause spasm of the pyloric sphincter and projectile vomiting. Vomiting in hyperammonemia is more likely mediated by the central nervous system.
Vomiting in infants with IEM does not occur without other symptoms such as lethargy, hypotonia or hypertonia, seizures, or coma.
Sepsis and gastroenteritis with dehydration also can have this constellation of symptoms, so maintain a high index of suspicion for these conditions when evaluating patients with suspected inborn errors of metabolism.46 Associated features of metabolic acidosis, hypoglycemia, elevated lactate or pyruvate, hyperammonemia, the presence or absence of ketosis, and a thorough family history, including possible consanguinity, can help elucidate the correct diagnosis.
Diarrhea, prominent in galactosemia and variably present in other disorders, is much less frequent than vomiting.6
Jaundice and Liver Dysfunction. Jaundice or other evidence of liver dysfunction may be the presenting finding in a number of inherited metabolic disorders in infancy.2,9 For most of the inborn errors of metabolism associated with jaundice, the elevated serum bilirubin is of the direct-reacting type. This generalization does not include those inborn errors of erythrocyte metabolism, such as glucose-6-phosphate dehydrogenase deficiency or pyruvate kinase deficiency, that are occasionally responsible for hemolytic disease in the newborn.
The best known metabolic disease associated with jaundice is galactosemia. In galactosemia a deficiency of the enzyme galactose-1-phosphate uridyl transferase, an enzyme important in the conversion of galactose (milk sugar) to glucose, results in an accumulation of galactose-1-phosphate and other metabolites, such as galactitol, that are thought to have a direct toxic effect on the liver and on other organs.9 Jaundice and liver dysfunction in this disorder are progressive and usually appear at the end of the first or during the second week of life, with vomiting, diarrhea, poor weight gain, and eventually cataract formation.5,7 Hypoglycemia may be observed. The disease may present initially with indirect hyperbilirubinemia resulting from hemolysis secondary to high levels of galactose-1-phosphate in erythrocytes. Alternatively, the effects of acute galactose toxicity on the brain may cause the CNS symptoms to predominate.
If the diagnosis of galactosemia is suspected, whether or not reducing substances are found in the urine, galactose-containing feedings should be discontinued immediately and replaced by soy formula or other lactose-free formula pending the results of an appropriate enzyme assay on erythrocytes to confirm the diagnosis. Untreated infants with galactosemia, if they survive the neonatal period, have persistent liver disease, cataracts, and severe mental retardation. Many affected infants die of Escherichia coli sepsis in the neonatal period, and the early onset of sepsis may alter the presentation of the disorder.5,7 Although many states have newborn screening programs for galactosemia, clinical manifestations of the disorder may appear before the results of screening studies are available, and it is therefore critical that physicians remain alert to this possibility.
Abnormal Odor. Abnormal body or urinary odor is an important, but often overlooked, clue to the diagnosis of several of the inborn errors of metabolism and may be the most specific clinical finding in these patients.9,14,47 In the acutely ill infant with an abnormal odor, isovaleric acidemia, glutaric acidemia type II, and maple syrup urine disease are the most likely entities to be encountered. In MSUD, the urine has a distinctive sweet odor, said to be reminiscent of maple syrup or burnt sugar. The odor associated with isovaleric acidemia and glutaric acidemia type II is pungent and unpleasant and similar to that of sweaty feet.
Emergency Treatment of the Infant With an Acute Metabolic Encephalopathy. When an inborn error of metabolism, such as an organic acidemia or urea cycle defect, is suspected in a critically ill infant, immediate treatment should be initiated, even if a definitive diagnosis may not yet be established. Within 48-72 hours, the results of amino acid and organic acid analysis should be available, allowing diagnostic confirmation in most cases. Appropriate and aggressive treatment before the confirmation of a diagnosis may be life-saving and may avert or reduce the neurologic sequelae of some of these disorders.
The key to emergency treatment of the critically ill child is to reverse the catabolic metabolic state to an anabolic state by administering dextrose-electrolyte intravenous solutions while supporting and monitoring the airway, breathing, and circulation.2,5 Since infection can mimic or accompany an IEM, obtain appropriate cultures and begin broad spectrum antibiotics.
An immediate treatment goal is the removal of accumulating metabolites such as organic acid intermediates or ammonia. At the first suspicion of a disorder associated with protein intolerance, protein intake should be discontinued immediately. In critically ill infants with hyperammonemia, arrangements should be made for hemodialysis. Although peritoneal dialysis, continuous arteriovenous hemoperfusion, and exchange transfusion all have been used in the past to lower plasma ammonia levels, all are substantially less effective than hemodialysis.35 In infants who are comatose or ventilator-dependent, or who exhibit evidence of cerebral edema, dialysis should be instituted immediately without waiting to determine whether there is a response to dietary manipulation, medication, or other less aggressive therapy.
If acidosis exists, intravenous bicarbonate should be administered liberally.4 Calculations of bicarbonate requirements appropriate for the treatment of other conditions are rarely adequate in these disorders because of ongoing production of organic acids or lactate. The acid-base status should be monitored frequently, with therapy adjusted accordingly. Dialysis should be considered for severely acidotic neonates with organic acidemias, regardless of whether hyperammonemia is present.4
In patients suspected of having a urea cycle defect because of significant hyperammonemia without acidosis, an infusion of 6 mL/kg of 10% arginine HCL (0.6 g/kg) can be given intravenously over 90 minutes.23 In patients with citrullinemia and argininosuccinic aciduria, this often results in a precipitous drop in the plasma ammonia level. An intravenous arginine preparation is available commercially and should be readily accessible to any hospital pharmacy.
If an organic acidemia is suspected, vitamin B12 (1 mg) should be given intramuscularly in case the patient turns out to have a B12-responsive form of methylmalonic acidemia.4 Biotin (10 mg) should be given orally or by nasogastric tube, because some patients with multiple carboxylase deficiency are biotin-responsive.
Samples to Obtain From a Dying Child with a Suspected Inborn Error of Metabolism. If death appears imminent in a child suspected of having an IEM, it is important to obtain the appropriate samples for postmortem analysis.2,8,48 This is critical for resolution of the cause of death and is essential for subsequent genetic counseling and prenatal diagnosis. The following samples should be collected and stored: urine, frozen; plasma, separated from whole blood and frozen; and a small snip of skin obtained using sterile technique and stored at room temperature or 37°C in tissue culture medium, if available, or sterile saline.7,48 If an autopsy is performed, a sample of unfixed liver tissue should be obtained as soon as possible after death and frozen at -20°C for subsequent biochemical studies.7 Additional tissue should be preserved for electron microscopy. If consent for autopsy is denied, consent for a postmortem needle biopsy of the liver can be requested. The liver tissue should be frozen in total or in part if histologic studies appear to be indicated. Whether before death or as soon as possible after death, the case should be reviewed with a metabolic specialist and plans made for the transport of samples to the appropriate laboratory.
Conclusion
Advances in diagnosis and treatment have improved the prognosis for many infants with IEM. Early clinical diagnosis is essential in ensuring that affected infants will receive the benefits of these advances. It is hoped that the information presented will assist the physician in the recognition and initial evaluation and stabilization of infants who may have an IEM. Finally, we offer these key summary points:
• A plasma ammonia level should be obtained for any child with unexplained encephalopathy.
• The presence of urine ketones in the neonatal period is suggestive of an organic acidemia.
• Metabolic acidosis with hyperammonemia and ketotic hypoglycemia suggests an organic acidopathy.
• The triad of encephalopathy, respiratory alkalosis and hyperammonemia characterize urea cycle disorders.
• Nonketotic hypoglycemia is suggestive of a defect in fatty acid oxidation.
References
1. Calvo M, Artuch R, Macia E, et al. Diagnostic approach to inborn errors of metabolism in an emergency unit. Pediatr Emerg Care 2000;16:405-408.
2. Burton BK. Inborn errors of metabolism in infancy: A guide to diagnosis. Pediatrics 1998;102:e69.
3. Fong C. Principles of inborn errors of metabolism: An exercise. Pediatr Rev 1995;16:390-395.
4. Goodman SI, Greene CL. Metabolic disorders of the newborn. Pediatr Rev 1994;15:359-365.
5. Seashore MR, Rinaldo P. Metabolic disease of the neonate and young infant. Semin Perinatol 1993;17:318-329.
6. Berry GT, Bennett MJ. A focused approach to diagnosing inborn errors of metabolism. Contemp Pediatr 1998;15:79-102.
7. Leonard JV. The early detection and management of inborn errors presenting acutely in the neonatal period. Eur J Pediatr 1985;143:253-257.
8. Ward JC. Inborn errors of metabolism of acute onset in infancy. Pediatr Rev 1990;11:205-216.
9. Barness LA. Analyzing signs and symptoms of metabolic diseases. South Med J 1996;89:163-166.
10. Lindor NM, Karnes PJ. Initial assessment of infants and children with suspected inborn errors of metabolism. Mayo Clin Proc 1995;70:987-988.
11. Chaves-Carballo E. Detection of inherited neurometabolic disorders: A practical clinical approach. Pediatr Clin North Am 1992;39:801-819.
12. Waber L. Inborn errors of metabolism. Pediatr Ann 1990;19:105-118.
13. Winter SC. Diagnosing metabolic disorders: One step at a time. Contemp Pediatr 1993;10:35-63.
14. Burlina AB, Bonafe L, Zacchello F. Clinical and biochemical approach to the neonate with a suspected inborn error of amino acid and organic acid metabolism. Semin Perinatol 1999;23:162-173.
15. Arens R, Gozal D, Williams JL, et al. Recurrent apparent life-threatening events during infancy: A manifestation of inborn errors of metabolism. J Pediatr 1993;123:415-418.
16. Harpey JP, Charpentier C, Paturneau-Jouas M. Sudden infant death syndrome and inherited disorders of fatty acid beta-oxidation. Biol Neonate 1990;58(suppl 1):70-80.
17. Dixon MA, Leonard JV. Intercurrent illness in inborn errors of intermediary metabolism. Arch Dis Child 1992;67:1387-1391.
18. American Academy of Pediatrics, Newborn Screening Task Force. A call for a national agenda on state newborn screening programs. Pediatrics 2000; 106(suppl):389-427.
19. Council of Regional Networks for Genetic Services. US newborn screening system guidelines II: Follow-up of children, diagnosis, management, and evaluation. J Pediatr 2000;137:S1-S46.
20. Morris AM, Hoffmann GF, Naughten ER, et al. Glutaric aciduria and suspected child abuse. Arch Dis Child 1999;80:404-405.
21. Hartley LM, Khwaja OS, Verity CM. Glutaric aciduria type 1 and non-accidental head injury. Pediatrics 2001;107:174-176.
22. Smith WE, Millington DS, Koeberl DD, et al. Glutaric acidemia, type I, missed by newborn screening in an infant with dystonia following promethazine administration. Pediatrics 2001;107:1184-1187.
23. Brusilow SW, Maestri NE. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv Pediatr 1996;43:127-169.
24. Reid SR. Hypoglycemia in infants and children. Pediatric Emergency Medicine Reports 2000;5:23-30.
25. Cornblath M, Hawdon JM, Williams AF, et al. Controversies regarding definition of neonatal hypoglycemia: Suggested operational thresholds. Pediatrics 2000;105:1141-1145.
26. Ichord RN. Perinatal metabolic encephalopathies. In: Swaiman KF, Ashwal S, eds. Pediatric Neurology: Principles and Practice. St. Louis: Mosby; 1999:220-233.
27. Cornblath M, Ichord R. Hypoglycemia in the neonate. Semin Perinatol 2000;24:136-149.
28. Venugopalan P, Agarwal AK, Worthing EA. Chronic cardiac failure in children due to dilated cardiomyopathy: Diagnostic approach, pathophysiology and management. Eur J Pediatr 2000;159:803-810.
29. Treem WR. New developments in the pathophysiology, clinical spectrum, and diagnosis of disorders of fatty acid oxidation. Curr Opin Pediatr 2000; 12:463-468.
30. Hale DE, Bennett MJ. Fatty acid oxidation disorders: A new class of metabolic diseases. J Pediatr 1992;121:1-11.
31. Stanley CA, Hale DE. Genetic disorders of mitochondrial fatty acid oxidation. Curr Opin Pediatr 1994;6:476-481.
32. Casteels-Van Daele M, Van Geet C, Wouters C, et al. Reye syndrome revisited: A descriptive term covering a group of heterogeneous disorders. Eur J Pediatr 2000;159:641-648.
33. Hostetler MA, Arnold GL, Mooney R, et al. Hypoketotic hypoglycemic coma in a 21 month old child. Ann Emerg Med 1999;34:394-398.
34. Msall M, Batshaw ML, Suss R, et al. Neurologic outcome in children with inborn errors of urea synthesis. N Engl J Med 1984;310:1500-1505.
35. Urea Cycle Disorders Conference Group. Consensus statement from a conference for the management of patients with urea cycle disorders. J Pediatr 2001;138(suppl):S1-S5.
36. Wraith JE. Ornithine carbamoyl transferase deficiency. Arch Dis Child 2001;84:84-88.
37. McEwan P, Simpson D, Kirk JM, et al. Hyperammonemia in critically ill septic patients. Arch Dis Child 2001;84:512-513.
38. Butterworth RF. Effects of hyperammonemia on brain function. J Inherit Metab Dis 1998;21(suppl 1):6-20.
39. Swenson ER. Metabolic acidosis. Respir Care 2001;46:342-353.
40. Thompson GN. Inborn errors of propionate metabolism: methylmalonic and propionic acidemias. J Paediatr Child Health 1992;28:134-135.
41. Breningstall GN. Approach to diagnosis of oxidative metabolism disorders. Pediatr Neurol 1993;9:81-90.
42. Tsao CY, Mendell JR, Lo WD, et al. Mitochondrial respiratory-chain defects presenting as nonspecific features in children. J Child Neurol 2000;15: 445-448.
43. Munnich A, Rotig A, Chretien D, et al. Clinical presentations and laboratory investigations in respiratory chain deficiency. Eur J Pediatr 1996;155: 262-274.
44. Sue CM, Hirano M, DiMauro S, et al. Neonatal presentations of mitochondrial metabolic disorders. Semin Perinatol 1999;23:113-124.
45. Evans OB. Symptomatic seizures. Pediatr Ann 1999;28:231-237.
46. Murray KF, Christie DL. Vomiting in infancy: When should you worry? Contemp Pediatr 2000;17:81-115.
47. Rizzo WB, Roth KS. On being led by the nose: Rapid detection of inborn errors in metabolism. Arch Pediatr Adolesc Med 1994;148:869-872.
48. Steiner RD, Cederbaum SD. Laboratory evaluation of urea cycle disorders. J Pediatr 2001;138:S21-S29.
49. Levy PA, Shapira E. Heterogeneity and variability of inborn errors of metabolism. AJDC 1993;147:265-266.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.