Meningococcal Disease Part I: Epidemiology, Etiology, Pathophysiology, and Clinical Features
Meningococcal Disease Part I: Epidemiology, Etiology, Pathophysiology, and Clinical Features
Authors: Sharon G. Humiston, MD, MPH, Associate Professor of Emergency Medicine, Department of Emergency Medicine, University of Rochester, NY; and Anne F. Brayer, MD, Department of Emergency Medicine, University of Rochester, NY.
Peer Reviewers: Robert W. Schafermeyer, MD, FACEP, FAAP, FIFEM, Associate Chair, Department of Emergency Medicine, Carolinas Medical Center; Adjunct Professor of Emergency Medicine and Pediatrics, University of North Carolina School of Medicine, Charlotte; and Ann Dietrich, MD, FAAP, FACEP, Associate Clinical Professor, Ohio State University, Attending Physician, Columbus Children's Hospital, Associate Pediatric Medical Director, MedFlight, Columbus, OH.
This issue of Emergency Medicine Reports begins the first in a two-part series on meningococcal disease. Part I of this series will focus on epidemiology, etiology, pathophysiology, and clinical features. Part II will follow with coverage of diagnostic studies, differential diagnosis, and management of the disease.
The Editor
Introduction
Neisseria meningitidis has the dubious distinction of being the last remaining serious bacterial threat to the lives and well being of otherwise healthy Americans. In industrialized nations, the risk of death or serious illness from organisms such as Haemophilus influenzae and Streptococcus pneumoniae has been reduced greatly by the development of vaccines against these encapsulated organisms.1-4 Newer vaccines soon will be available against some strains of the meningococcus,5,6 but until they achieve widespread effectiveness and availability, meningococcal disease will continue to be a significant cause of morbidity and mortality in the population. The meningococcus causes a variety of disease entities, but this review focuses chiefly on its two major manifestations: severe meningococcal septicemia (sometimes confusingly called "meningococcemia") and meningitis. One possible reason for the relative lack of reduction in mortality from severe sepsis with this organism may be the failure of clinicians to understand the pathophysiologic differences between these two, and their implications for emergency management.7,8
Relevance to the Emergency Department Population
Prompt recognition and treatment of meningococcal disease in the emergency department (ED) can be literally life- and limb-saving, while missed clues can produce devastating and largely preventable consequences.9,10 The rapid progression of illness means that once the patient's condition has become apparent, resuscitation and stabilization will require the particular skills and resources only available in an ED setting. In addition, several of the risk factors for meningococcal disease, such as low socio-economic status and adolescent or young adult age, also are associated with frequent use of ED rather than of primary care services.11,12
Epidemiology
As many as 3000 people in the United States experience meningococcal disease each year.2 While outbreaks of illness tend to receive major attention from the media and the general public, fewer than 5% of cases actually occur during outbreaks.13,14 The prevalence of the asymptomatic carrier state varies from less than 2% in children younger than 2 years of age to as high as 10-40% among adolescents and young adults.15-17 The highest carrier prevalence is found among those living in close quarters, such as college students and military recruits.
Actual disease incidence rates range between 0.9 and 1.5 per 100,000 in the general population and have not changed significantly in the past 40 years.2,18 Incidence rates are as high as 5 per 100,000 among infants, declining steadily through late school age, but with an additional peak in the adolescent and young adult years (incidence rate in 2003 was 0.63 in 11-17 year-olds and 1.0 in 18-22 year-olds).14 There also is seasonal variation in incidence, with peak occurrences in late winter and spring.18
Case fatality rates vary between 8% and 13%.14 Fatality rates generally increase with age in the populations with highest prevalence;19,20 12% of 10-17 year-olds and 22.5% 15-24 year-olds died in two recent surveillance studies.14,18 Survivors sustain considerable long-term morbidity, with 12-19% having sequelae such as hearing loss, brain injury, or amputations.21,22
Etiology
The interplay of multiple factors determines which individuals will become ill with a particular organism. These factors traditionally are classified as attributes of the organism, the host, and the environment. The development of invasive meningococcal disease is a classic example of the interplay among these, with a relatively high prevalence of a fairly virulent organism causing disease in a relatively small number of victims.
Causative FactorsThe Organism. The causative agent of meningococcal disease is the aerobic Gram-negative diplococcus Neisseria meningitidis, which is a natural comensal organism in the nasopharynx of humans, its only host.23 The relevant components of the organism are its outer membrane and the polysaccharide capsule. The membrane is the site of the lipopolysaccharide molecule, the endotoxin that triggers the pathological immune response (see below).20 Capsular polysaccharide composition varies, and it is on this basis that the 13 distinctive serogroups of the organism are identified. Virtually all invasive disease is caused by meningococci in one of the five serogroups A, B, C, Y, and W-135. Groups B, C, and Y cause the bulk of disease in North America, while groups A and C predominate in Asia and Africa.20,24 W-135 has been responsible for sporadic outbreaks in Africa and the Middle East, including one well-documented recent outbreak among pilgrims returning from the Hajj.25 Micropilli, or fimbriae on the outer surface of the capsule, are the basis for adhesion to the nasopharyngeal epithelium.26 Colonization with the organism is the most common outcome of adhesion, and is an immunizing event for the host producing asymptomatic carriage.2 Systemic infection occurs in fewer than 1% of asymptomatic carriers when changes in the mucosal barrier or host immune factors permit invasion of the bloodstream.27
Predisposing ConditionsThe Host. Age already has been mentioned as a predisposing factor, with children younger than 2 years of age experiencing nearly a five-fold increase in risk over that in the general adult population. Defects in the host defense mechanisms, both congenital and acquired, predispose to bacterial invasion and active disease. Patients with sickle cell anemia are functionally splenectomized after the age of about 2 years, which produces a defect in their ability to clear encapsulated organisms in general. Such patients suffer a disproportionate burden of illness severity, morbidity, and mortality from meningococcal disease. In otherwise healthy hosts, acute viral respiratory infections are thought to be predisposing factors.28-30 Because invasion by the organism triggers a response from virtually every branch of the host immune system (see below), almost all immune deficiency states are predisposing factors for disease.23,27,31 Stephens reported that two-thirds of adults with meningococcal disease had one or more immunocompromising conditions.32 The complement system is of major importance in host defense against invasion by bacterial organisms, and complement deficiency states are important risk factors.31 There is growing evidence that specific genetic polymorphisms also may underlie increased risk for invasive meningococcal disease, presumably by altering the structure or function of immune system components.33-36
Risk FactorsThe Environment. Environmental factors that affect individual risk of invasive disease can be divided into those factors that promote person-to-person spread of the organism and thus colonization, and those that affect the function of the nasopharyngeal mucosal barrier, thereby facilitating invasion of the bloodstream. Crowded living conditions are perhaps the best-known circumstances that predispose to transmission of the organism. The rate of secondary infection among household contacts is up to 800 times that in the general population.2 Transmission occurs by short-range exposure (e.g., droplet aerosolization or direct contact with secretions) and thus, effective transmission requires close person-to-person contact. Not surprisingly, this condition is met by members of most of the adult at-risk populations: dormitory-dwelling college students,37 military recruits,38 and people who live in socioeconomically deprived geographic areas.12,39 It seems likely that the latter factor is a surrogate marker for household crowding and other environmental factors such as tobacco smoke and high rates of respiratory viral infection.40 Psychosocial stressors associated with deprivation, such as frequent moves, household arguments, and legal disputes were associated significantly and independently with elevated risk in one study, although it was unclear how this association might be mediated.41
Both active and passive exposure to tobacco smoke greatly increases the risk of illness through disruption of the mucosal barrier26 and by a variety of immunosuppressive effects.42,43 It also contributes to increased transmission of the organism by increasing production of respiratory droplets.44 The risk of invasive bacterial disease in general among adults is increased by two- to four-fold in smokers.4,43,45 Asymptomatic carriage rates are substantially higher among smokers.15 Children, who already experience the highest rates of illness, see even higher relative risks with smoke exposure, estimated at 3.5 to 7.5 times that in the general population.41,46,47 There appears to be a positive dose-response relationship between passive smoke exposure and risk.48 Of significance, day-care attendance actually may reduce the risk of invasive disease among children who live with smokers, possibly by reducing the amount of time the young child spends in close contact with multiple adults who are asymptomatic carriers.49
Pathophysiology
Mechanism of Disease Process. N. meningitidis produces a variety of disease manifestations (see Table 1), but the two most common and devastating are meningococcal meningitis (MM) and severe meningococcal sepsis (SMS), discussed separately below. There are two critical events in the pathogenesis of meningococcal disease: penetration of the organism through the nasopharyngeal mucosa and replication in the bloodstream. Invasive disease occurs only after penetration, and once penetration does occur the response to replication defines the course. If replication is rapid and overwhelms host defenses, SMS is the result, whereas if replication can be held in partial check by immune mechanisms, localizing disease such as meningitis or other suppurative complications develop. The reasons why some individuals develop SMS while others develop MM are not clear.23
| Table 1. Meningococcal Disease—Infectious Syndromes* |
|
|

Colonization and Invasion. Following exposure, the organism colonizes the host nasopharynx (NP). (See Figure 1.) N. meningitidis produces virulence factors that include the polysaccharide capsule and its associated structures that promote adhesion to mucosal cells,50 proteases that destroy host secretory (IgA) antibodies,51 and mucosal ciliary inhibitors.17,52 Hosts with fully functioning immune systems typically either destroy the organism shortly after exposure or, at worst, establish an asymptomatic carrier state. In either case, humoral immunity is produced.2 In hosts with impaired mucosal barriers (e.g., smokers, those with acute viral illnesses), or with immunocompromise, invasion through the mucosa, survival in the bloodstream, and rapid multiplication of the organism set the stage for severe disease.23
| Figure 1. Colonization and Invasion by N. Meningitidis |
|
|
| Copyright 2001 Massachusetts Medical Society. Reprinted with permission from: Rosenstein NE, Perkins BA, Stephens DS, et al. Meningococcal disease. N Engl J Med 2001;344:1383. |
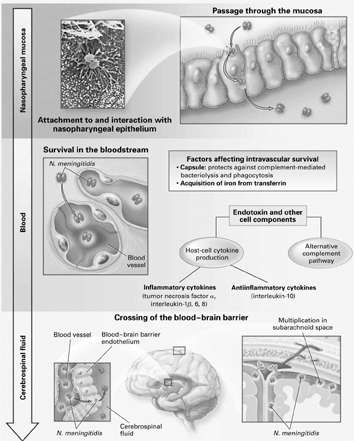
Virtually every branch of the human immune system is involved in response to penetration of the organism into the bloodstream. Until the maturation of an individual's ability to mount an acquired immune response (after the first year of life), the innate immune system, particularly complement, provides the main defense against N. meningitidis. This explains the high peak of incidence during late infancy, as passive immunity provided by maternal antibody subsides. Acquisition of both antibody- and cell-mediated immunity occurs throughout childhood, accounting for the drop in incidence during that period.23
The Inflammatory Response and Microvascular Injury. The host inflammatory immune response that follows penetration of primary defenses is intimately involved in the pathogenesis and clinical manifestations of meningococcal disease. Indeed, a recent review suggests that it can be useful to view meningococcal sepsis as an inflammatory disease.53 Many of the triggers of the immune/inflammatory response to the meningococcus are so-called "pattern recognition receptors" that identify molecular structures common to many pathogens.54 These receptors are less specific at distinguishing self from non-self antigens than are the components of the innate immune system, which may account for the profound damage done to host tissues.55
Pattern recognition receptors mediate both phagocytosis and activation of pro-inflammatory cytokine pathways.56,57 The cytokine contribution to the inflammatory cascade must be maintained in a delicate balance so that sufficient response to pathogens occurs while minimizing damage to host tissues.53 Plasma concentrations of pro-inflammatory cytokines such as tumor necrosis factor (TNF) and various interleukins (IL) increase dramatically during acute infection with meningococcus,58-60 although the causal meaning of this observation is unclear. The interaction between pro- and anti-inflammatory cytokines has been postulated to be related to the clinical manifestation of disease (e.g., SMS vs. meningitis).61
The ultimate result of the activation of the cytokine-mediated inflammatory cascade in meningococcal disease is an assault on the capillary endothelium of the host.62,63 Virtually every sign and symptom, most newer therapies, and many prognostic indicators hinge on the immune-mediated injury to vascular endothelial cells.23 The multifactorial immune-mediated microvascular injury produces the four general manifestations of pathology caused by meningococcal infection: capillary leak, vasomotor instability, disordered coagulation, and myocardial dysfunction.23,64 These, in turn, account for the various multiple organ and system failures that are observed in clinical disease. (See Table 2.)
| Table 2. Causes of Intravascular Thrombosis in Meningococcal Sepsis |
|
|

Specific Organ Systems Involved. The specific result of the four general manifestations of capillary damage is impairment and ultimately failure of most of the major organ systems.22,23,26,65,66
The cardiovascular system profoundly is affected. Myocardial function is impaired in SMS, with decreased stroke volume resulting in diminished cardiac output.67 In children, diminished stroke volume can be transiently compensated for by increased heart rate, but this comes at the expense of increased metabolic demand. Myocardial ischemia and cell death with elevation of serum levels of troponin I occur in patients with SMS68 and are correlated with the degree of myocardial dysfunction.69 The pro-inflammatory cytokines TNF-alpha and IL-1beta are known to reduce myocyte contractility in vitro.70 There is evidence that the ultimate mediator of reduced contractility may be nitrous oxide and cyclic guanosine monophosphate (GMP) induced by the elevated cytokine levels.71
Central nervous system (CNS) impairment occurs by two quite distinct mechanisms, which may be present alone or in combination in any given patient. In MM, inflammatory changes to vascular permeability and the blood-brain barrier, as well as polymorphonuclear infiltrates, produce the clinical picture of meningitis and direct inflammation of brain.26,72 Increased cerebrospinal fluid (CSF) production and decreased reabsorption, along with cerebral edema, produce rapidly elevated intracranial pressure (ICP). These changes result in diminished consciousness, confusion, and ultimately respiratory compromise if brain herniation occurs.73,74 By contrast, patients with SMS who are in rapidly progressive shock will experience reduced perfusion, tissue acidosis, and ultimately cerebral infarctionend-organ effects similar to those produced in other body systems by massive circulatory compromise.
The characteristic evolving rash of meningococcal disease is the result of damage to capillaries and the endothelium of small end-arteries.75,76 Vasculitis with extravasation of red blood cells (and viable organisms) from leaking capillaries produces the initial petechial exanthem.7 With progression, micro- and macroscopic thrombi form in end-arteries and arterioles, producing varying degrees of ischemia and ultimately necrosis and gangrenous changes if perfusion is not restored.65,77
Other organs that notably are affected by microvascular injury and its consequences include the lungs, where capillary leak and infiltration with neutrophils produce both intra-alveolar fluid and thickening of the pulmonary interstium.78 These changes reduce alveolar gas exchange and contribute to the "stiff lung" of adult respiratory distress syndrome (ARDS). This leads to initial tachypnea followed by frank respiratory failure with pulmonary edema. There is a single report of the development of ascites of sufficient quantity to compromise lung volume.79
Renal blood flow suffers during SMS in direct proportion to the degree of shock.80 This effect may be exaggerated in younger individuals.81 Oliguria or anuria may follow, and in severe cases permanent kidney damage from acute tubular necrosis may occur.82
Splanchnic blood flow in general is reduced, and thrombi forming in the mesenteric or gastric distributions can produce submucosal ischemia and hemorrhage similar to what is seen on skin; some patients may complain of severe abdominal pain.83 In rare cases these lesions may erode and form ulcers.84
Vascular injury also contributes to hepatocellular damage, infarction, and hemorrhage of the adrenal glands (Waterhouse-Friderichsen Syndrome) and virtually every other organ and system.85,86
Clinical Features
Communication about disease caused by N. meningitidis is hampered by confusing and often conflicting terminology.8,87 Only about half of patients with meningococcal bacteremia (that is, whose blood cultures grow the organism) actually have isolated meningitis (purulent inflammation of the meninges with accompanying cerebro-encephalitis).22 It is best to use the term severe meningococcal sepsis or SMS to refer to the systemic manifestations of the organism reproducing in the blood as an organ, and to differentiate this rapidly progressive septic state from localized (though still potentially serious) disease. Ten to fifteen percent of patients have SMS alone,19,20 and 40% typically have a mixed picture.7,22 Too often, clinicians associate petechiae or purpura only with meningitis and waste valuable time diagnosing and managing it, when the real culprit, fulminant sepsis, progresses rapidly and without adequate notice.87 The use of the term "meningococcemia," can be confusing and can contribute to delays in treatment88 (though unfortunately the term still is widely used both in the literature and clinically).
Major Clinical Syndromes. Severe Meningococcal Sepsis (SMS). SMS is characterized by sudden onset, rapid progression, and an absence of localizing findings. The presentation usually is more severe than in meningitis or other manifestations. Most patients with SMS have no known immunocompromise. These features of SMS likely account in large part for its much higher case fatality rate (40-50%).7,8
Presenting Symptoms and Physical Examination. Meningococcal septicemia begins with an acute onset of high fever, shaking chills, and myalgias that may be expressed as extremity pain89 or back pain,8 particularly in adolescents or young adults. Patients presenting to the ED with these symptoms before the onset of a rash are at high risk for being sent home with a diagnosis of "viral syndrome,"90 especially because there often is a transient improvement within six hours of onset.8
Within another six hours, however, patients with SMS invariably deteriorate rapidly, most commonly with development of a rash that initially may resemble a viral exanthem,8 although it is more classically petechial in character. The rash becomes hemorrhagic, and most commonly coalesces to form widespread purpuric lesions. Purpura fulminans, or aggressive spread of purpura to large areas with ischemic necrosis, may develop. Patients with purpura fulminans are likely to have sudden drops in blood pressure and acute adrenal hemorrhage (Waterhouse-Friderichsen Syndrome).86
Vital Signs. Early SMS can present with normal blood pressure and warm extremities, and especially in children, tachycardia may be the only firm sign of impending disaster.7 Vital signs other than temperature often initially are reported as "normal," though with disturbing frequency post-mortem chart reviews reveal evident abnormalities such as elevated heart rate for age and temperature, or widened pulse pressure. Normal blood pressure does not signal reassurance.91 Altered mental status ranging from anxiety, confusion, and combativeness to lethargy and stupor may be early manifestations of poor brain perfusion.23
Clinical Clues to Early Diagnosis of SMS. One of the reasons that mortality rates have remained unchanged for four decades is that this early period usually represents the only window for effective intervention, and early diagnosis and treatment continue to evade clinicians.92 Yung and McDonald recently have published a set of clinical pearls which, while somewhat general, may help in the early recognition of meningococcal sepsis.8 (See Table 3.)
| Table 3. Common Signs and Symptoms of Meningococcal Disease |
|
|

Skin Findings. Any rash appearing in the context of a sudden febrile illness should raise concern. Unlike viral syndromes that have a several-day prodrome before development of the rash, in patients with meningococcal disease the rash typically is present within the first 24 hours of any symptomatology. The first petechiae may be intraoral, conjunctival, or be hidden in skin folds of the axilla, groin, or other regions. Notably, the early rash may not be petechial or hemorrhagic at allit may be diffuse and maculopapular90 and may blanch with pressurea falsely reassuring finding.
Rigors. Yung and McDonald suggest that the presence of "true rigors," that is, a prolonged (10-20 minute) shaking chill that cannot be stopped voluntarily, is a strong indication of sepsis.8 These authors recommend admission for antibiotics and observation of any febrile patient with true rigors. The absence of true rigors should not be completely reassuring, however.
Localized Pain. Localized extremity or muscle pain also is cited as something sufficiently unusual in the typical febrile patient to warrant concern, particularly in older children.93 Younger children may refuse to walk.89 Similarly, abdominal pain, back pain, and/or vomiting in a previously healthy febrile patient, absent diarrhea, is sufficiently unusual to merit a closer look. Yung and MacDonald recommend paying "a great deal of attention to any febrile patient with severe pain at any site."8
Patient and Family Characteristics. Yung also appropriately points out that because previously healthy young people rarely seek care abruptly, a sudden change in health should be taken very seriously, as should a patient, parent, or friends who seem more concerned than the objective signs might suggest.8 This recommendation has been supported in a recent qualitative study.94
Yung concludes the excellent clinical review with two important observations: 1) Fever and a petechial or hemorrhagic rash is always SMS until proven otherwise; and 2) while no single finding is an indication for immediate treatment and admission, one always should give serious consideration to meningococcal disease when one or more of the signs are present.8 Ask, "Why is this patient seeking help right now for this problem?" When in doubt, use aggressive fluid management and early administration of antibiotics.
Bear in mind that in a typical year most emergency medicine providers will see hundreds of children with many of the symptoms presented in Table 3; most will turn out to have a simple viral syndrome. It is the presence of several or many of these findings in a previously well patient, and their rapid progression, that should trigger the clinical alarm, not their individual occurrence.
Meningococcal Meningitis (MM). In the 50% of patients with meningitis caused by N. meningitidis but without fulminant sepsis, signs and symptoms are those of typical bacterial meningitis. Although they do not have the end-organ manifestations of SMS, these patients are (or have been) bacteremic with the organism, and progression to sepsis and shock is an ever-present possibility.
Presenting Symptoms and Physical Examination. Patients with MM usually have a one- to three-day non-specific prodrome that resembles viral illness, with low-grade fever and upper respiratory symptoms.8 Myalgias, and especially back pain, are common. Signs and symptoms progress in adolescent and adult patients, classically, to sudden and severe headache, often with photophobia, and the development of a higher fever and stiff neck. Children and adult patients who are lucid may complain of worsening headache with neck flexion. Nausea and vomiting are common.20 Older children may complain of painful extremities,89 while infants may not present with meningismus or stiff neck.20
Patients may have mental status ranging from normal to obtunded. A petechial rash sometimes is present, and is not necessarily a sign of more severe disease.95 In patients older than 3 years, the classical meningeal signs of Kernig and Brudzinski may be elicited. [The Kernig sign is present when the supine patient with the thigh flexed onto the abdomen complains of pain on passive extension of the leg. The best known of Brudzinski's five meningeal signs is produced in the supine patient when passive neck flexion produces spontaneous flexion of the hips and knees.96] While useful if present, these signs rely on a cooperative patient with near-normal mental status. Infants and toddlers typically do not manifest the classical findings of meningeal inflammation. They may be lethargic or irritable. Younger infants may demonstrate a bulging fontanel, but its absence does not rule out meningitis.
In MM, direct cerebral inflammation and rising intracranial pressure (ICP) may produce the familiar signs of lethargy progressing to obtundation, accompanied by the Cushing triad of hypertension, bradycardia, and respiratory depression culminating in respiratory arrest. They also may have centrally mediated vasospasm resulting in decreased peripheral perfusion. Vital signs will show markedly different trends in the two cases, with SMS patients exhibiting tachycardia and (eventually) hypotension, and MM patients developing hypertension and bradycardia. When in doubt, support the circulating volume and observe the response.
Other Forms of Localized Meningococcal Disease. SMS and MM are the most common manifestations of invasive disease caused by N. meningitidis, but other localized forms of infection occur. As with MM, these conditions always are preceded by a bacteremic phase followed by seeding of the infected site. Although it is unusual in children, meningococcal pneumonia is found in up to 15% of adults with invasive disease97 and is more common in patients with immunodeficiency states.32 As with N. gonorrheae, meningococcus may cause conjunctivitis,98 urethritis,99 or arthritis.100 Because these syndromes present with slowly worsening disease and usually are recognized easily, mortality and long-term morbidity typically are fairly low with prompt and appropriate treatment.8
Chronic Meningococcemia. Patients with complement31 or other immunodeficiencies101 (and rarely those without) can develop chronic meningococcemia (in this instance, the term is both accurate and descriptive). In this condition, the organism circulates in blood without localizing and without the rapid reproduction and endotoxin release seen in SMS. Such patients almost never are diagnosed on their first encounter with the health care system, but present with intermittent fevers over a several-week period, accompanied by an evanescent non-petechial rash, arthralgias, and headache.20 These patients often undergo evaluation for Rickettsial diseases, Lyme disease, or other arthropod- or animal-borne infections before the correct diagnosis is reached.
References
1. Progress toward elimination of Haemophilus influenzae type b invasive disease among infants and childrenUnited States, 1998-2000. MMWR Morb Mortal Wkly Rep 2002 March 22;51:234-237.
2. National Foundation for Infectious Diseases. The Changing Epidemiology of Meningococcal Disease Among U.S. Children, Adolescents, and Young Adults. http://www.nfid.org/publications/meningococcalepid.pdf. (Accessed 4/10/2005): National Foundation for Infectious Diseases; 2004.
3. Whitney CG, Farley MM, Hadler J, et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med 2003;348:1737-1746.
4. Gold R. Epidemiology of bacterial meningitis. Infect Dis Clin North Am 1999;13;515-525, v.
5. Miller E, Salisbury D, Ramsay M. Planning, registration, and implementation of an immunisation campaign against meningococcal serogroup C disease in the UK: A success story. Vaccine 2001;20 Suppl 1:S58-67.
6. Soriano-Gabarro M, Stuart JM, Rosenstein NE. Vaccines for the prevention of meningococcal disease in children. Semin Pediatr Infect Dis 2002;13:182-189.
7. Welch SB, Nadel S. Treatment of meningococcal infection. Arch Dis Child 2003;88:608-614.
8. Yung AP, McDonald MI. Early clinical clues to meningococcaemia. Med J Aust 2003;178:134-137.
9. Kirsch EA, Giroir BP. Improving the outcome of septic shock in children. Curr Opin Infect Dis 2000;13:253-258.
10. Riordan FA. Improving promptness of antibiotic treatment in meningococcal disease. Emerg Med J 2001;18:162-163.
11. Harrison LH, Pass MA, Mendelsohn AB, et al. Invasive meningococcal disease in adolescents and young adults. JAMA 2001;286:694-699.
12. Williams CJ, Willocks LJ, Lake IR, et al. Geographic correlation between deprivation and risk of meningococcal disease: An ecological study. BMC Public Health 2004;4:30.
13. Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 2000;49(RR-7):1-10.
14. Centers for Disease Control and Prevention (CDC). Active Bacterial Core Surveillance (ABCs) 1997-2002 meningococcal surveillance reports. Available at www.cdc.gov/ncidod/dbmd/abcs/. Accessed 5/6/2005.
15. Block C, Gdalevich M, Buber R, et al. Factors associated with pharyngeal carriage of Neisseria meningitidis among Israel Defense Force personnel at the end of their compulsory service. Epidemiol Infect 1999;122:51-57.
16. Caugant DA, Hoiby EA, Magnus P, et al. Asymptomatic carriage of Neisseria meningitidis in a randomly sampled population. J Clin Microbiol 1994;32:323-330.
17. Stephens DS. Uncloaking the meningococcus: dynamics of carriage and disease. Lancet 1999;353:941-942.
18. Control and prevention of meningococcal disease: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 1997;46(RR-5):1-10.
19. Rosenstein NE, Perkins BA, Stephens DS, et al. The changing epidemiology of meningococcal disease in the United States, 1992-1996. J Infect Dis 1999;180:1894-1901.
20. Rosenstein NE, Perkins BA, Stephens DS, et al. Meningococcal disease. N Engl J Med 2001;344:1378-1388.
21. Edwards MS, Baker CJ. Complications and sequelae of meningococcal infections in children. J Pediatr 1981;99:540-545.
22. Kirsch EA, Barton RP, Kitchen L, et al. Pathophysiology, treatment and outcome of meningococcemia: A review and recent experience. Pediatr Infect Dis J 1996;15:967-978.
23. Pathan N, Faust SN, Levin M. Pathophysiology of meningococcal meningitis and septicaemia. Arch Dis Child 2003;88:601-617.
24. Riedo FX, Plikaytis BD, Broome CV. Epidemiology and prevention of meningococcal disease. Pediatr Infect Dis J 1995;14:643-657.
25. Centers for Disease Control and Prevention. Risk for meningococcal disease associated with the Hajj 2001. MMWR Morb Mortal Wkly Rep 2001;50:97-98.
26. Tunkel AR, Scheld WM. Pathogenesis and pathophysiology of bacterial meningitis. Clin Microbiol Rev 1993;6:118-136.
27. van DM, Brandtzaeg P, van der Meer JW. Update on meningococcal disease with emphasis on pathogenesis and clinical management. Clin Microbiol Rev 2000;13:144-166, table.
28. Cartwright KA, Jones DM, Smith AJ, et al. Influenza A and meningococcal disease. Lancet 1991;338:554-557.
29. Moore PS, Hierholzer J, DeWitt W, et al. Respiratory viruses and mycoplasma as cofactors for epidemic group A meningococcal meningitis. JAMA 1990;264:1271-1275.
30. Alonso JM, Taha MK. [Respiratory virosis and invasive bacterial superinfections. The case for influenza and meningococcal diseases]. Arch Pediatr 2003;10:1013-1015.
31. Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 1991;4:359-395.
32. Stephens DS, Hajjeh RA, Baughman WS, et al. Sporadic meningococcal disease in adults: Results of a 5-year population-based study. Ann Intern Med 1995;123:937-940.
33. Hibberd ML, Sumiya M, Summerfield JA, et al. Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Meningococcal Research Group. Lancet 1999;353:1049-1053.
34. Nadel S, Newport MJ, Booy R, et al. Variation in the tumor necrosis factor-alpha gene promoter region may be associated with death from meningococcal disease. J Infect Dis 1996;174:878-880.
35. Texereau J, Pene F, Chiche JD, et al. Importance of hemostatic gene polymorphisms for susceptibility to and outcome of severe sepsis. Crit Care Med 2004;32(5 Suppl):S313-S319.
36. Tzeng YL, Stephens DS. Epidemiology and pathogenesis of Neisseria meningitidis. Microbes Infect 2000;2:687-700.
37. Bruce MG, Rosenstein NE, Capparella JM, et al. Risk factors for meningococcal disease in college students. JAMA 2001;286:688-693.
38. Brundage JF, Ryan MA, Feighner BH, et al. Meningococcal disease among United States military service members in relation to routine uses of vaccines with different serogroup-specific components, 1964-1998. Clin Infect Dis 2002;35:1376-1381.
39. Haynes R, Gale S. Deprivation and poor health in rural areas: Inequalities hidden by averages. Health Place 2000;6:275-285.
40. Jackson LA, Wenger JD. Laboratory-based surveillance for meningococcal disease in selected areas, United States, 1989-1991. MMWR CDC Surveill Summ 1993;42:21-30.
41. Stanwell-Smith RE, Stuart JM, Hughes AO, et al. Smoking, the environment and meningococcal disease: A case control study. Epidemiol Infect 1994;112:315-328.
42. Haustein KO. [Health consequences of passive smoking]. Wien Med Wochenschr 2000;150:233-244.
43. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med 2004;164:2206-2216.
44. Fischer M, Hedberg K, Cardosi P, et al. Tobacco smoke as a risk factor for meningococcal disease. Pediatr Infect Dis J 1997;16:979-983.
45. McCall BJ, Neill AS, Young MM. Risk factors for invasive meningococcal disease in southern Queensland, 2000-2001. Intern Med J 2004;34:464-468.
46. Kriz P, Bobak M, Kriz B. Parental smoking, socioeconomic factors, and risk of invasive meningococcal disease in children: A population based case-control study. Arch Dis Child 2000;83:117-121.
47. Yusuf HR, Rochat RW, Baughman WS, et al. Maternal cigarette smoking and invasive meningococcal disease: A cohort study among young children in metropolitan Atlanta, 1989-1996. Am J Public Health 1999;89:712-717.
48. Pereiro I, ez-Domingo J, Segarra L, et al. Risk factors for invasive disease among children in Spain. J Infect 2004;48:320-329.
49. Grein T, O'Flanagan D. Day-care and meningococcal disease in young children. Epidemiol Infect 2001;127:435-441.
50. Plant L, Jonsson AB. Contacting the host: Insights and implications of pathogenic Neisseria cell interactions. Scand J Infect Dis 2003; 35:608-613.
51. Merz AJ, So M. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu Rev Cell Dev Biol 2000;16:423-457.
52. Stephens DS, Farley MM. Pathogenic events during infection of the human nasopharynx with Neisseria meningitidis and Haemophilus influenzae. Rev Infect Dis 1991;13:22-33.
53. Kvalsvig AJ, Unsworth DJ. The immunopathogenesis of meningococcal disease. J Clin Pathol 2003;56:417-422.
54. Jack DL, Dodds AW, Anwar N, et al. Activation of complement by mannose-binding lectin on isogenic mutants of Neisseria meningitidis serogroup B. J Immunol 1998;160:1346-1353.
55. Janeway CA, Jr. How the immune system works to protect the host from infection: A personal view. Proc Natl Acad Sci U S A 2001;98:7461-7468.
56. Jack DL, Jarvis GA, Booth CL, et al. Mannose-binding lectin accelerates complement activation and increases serum killing of Neisseria meningitidis serogroup C. J Infect Dis 2001;184:836-84 5.
57. Jack DL, Read RC, Tenner AJ, et al. Mannose-binding lectin regulates the inflammatory response of human professional phagocytes to Neisseria meningitidis serogroup B. J Infect Dis 2001;184:1152-1162.
58. Waage A, Brandtzaeg P, Halstensen A, et al. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J Exp Med 1989;169:333-338.
59. Riordan FA, Marzouk O, Thomson AP, et al. Pro-inflammatory and anti-inflammatory cytokines in meningococcal disease. Arch Dis Child 1996;75:453-454.
60. van Deuren M, Brandtzaeg P, van der Meer JW. Update on meningococcal disease with emphasis on pathogenesis and clinical management. Clin Microbiol Rev 2000;13:144-166, table.
61. van DM, van d, V, Vannier E, et al. The pattern of interleukin-1beta (IL-1beta) and its modulating agents IL-1 receptor antagonist and IL-1 soluble receptor type II in acute meningococcal infections. Blood 1997;90:1101-1108.
62. Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med 1993;328:1471-1477.
63. Hack CE, Zeerleder S. The endothelium in sepsis: Source of and a target for inflammation. Crit Care Med 2001;29(7 Suppl):S21-S27.
64. Baines PB, Hart CA. Severe meningococcal disease in childhood. Br J Anaesth 2003;90:72-83.
65. de Kleijn ED, Hazelzet JA, Kornelisse RF, et al. Pathophysiology of meningococcal sepsis in children. Eur J Pediatr 1998;157:869-880.
66. Williams AJ, Nadel S. Bacterial meningitis: Current controversies in approaches to treatment. CNS Drugs 2001;15:909-919.
67. Boucek MM, Boerth RC, Artman M, et al. Myocardial dysfunction in children with acute meningococcemia. J Pediatr 1984;105:538-542.
68. Thiru Y, Pathan N, Bignall S, et al. A myocardial cytotoxic process is involved in the cardiac dysfunction of meningococcal septic shock. Crit Care Med 2000;28:2979-2983.
69. Briassoulis G, Narlioglou M, Zavras N, et al. Myocardial injury in meningococcus-induced purpura fulminans in children. Intensive Care Med 2001;27:1073-1082.
70. Kumar A, Thota V, Dee L, et al. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med 1996;183:949-958.
71. Kumar A, Brar R, Wang P, et al. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol 1999;276(1 Pt 2):R265-R276.
72. Kim KS, Wass CA, Cross AS. Blood-brain barrier permeability during the development of experimental bacterial meningitis in the rat. Exp Neurol 1997;145:253-257.
73. Leppert D, Leib SL, Grygar C, et al. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: Association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis 2000;31:80-84.
74. Quagliarello V, Scheld WM. Bacterial meningitis: Pathogenesis, pathophysiology, and progress. N Engl J Med 1992;327:864-872.
75. Famularo G, Pozzessere C, Trinchieri V, et al. Fulminant purpuric rash. Eur J Emerg Med 2000;7:313-315.
76. Garcia-Porrua C, Gonzalez-Gay MA. Bacterial infection presenting as cutaneous vasculitis in adults. Clin Exp Rheumatol 1999;17:471-473.
77. Wharton SM, Reid CA. Purpura fulminans localising to a recent burn injury. Burns 1998;24:680-682.
78. Martin MA, Silverman HJ. Gram-negative sepsis and the adult respiratory distress syndrome. Clin Infect Dis 1992;14:1213-1228.
79. Beck R, Halberthal M, Zonis Z, et al. Abdominal compartment syndrome in children. Pediatr Crit Care Med 2001;2:51-56.
80. Brandtzaeg P, Sandset PM, Joo GB, et al. The quantitative association of plasma endotoxin, antithrombin, protein C, extrinsic pathway inhibitor and fibrinopeptide A in systemic meningococcal disease. Thromb Res 1989;55:459-470.
81. Hazelzet JA, Stubenitsky R, Petrov AB, et al. Cardiovascular aspects of experimental meningococcal sepsis in young and older awake piglets: Age-related differences. Shock 1999;12:145-154.
82. Shahidi-Asl M, Ananth M, Boineau F, et al. Apparent progression of acute glomerulonephritis to dense deposit disease. Ultrastruct Pathol 2000;24:273-277.
83. Lannon DA, Smyth YM, Waldron R. ‘Acute abdomen' with a rash. Int J Clin Pract 2000;54:470-471.
84. Britto J, Nadel S, Habibi P, et al. Gastrointestinal perforation complicating meningococcal disease. Pediatr Infect Dis J 1995;14:393-394.
85. Agraharkar M, Fahlen M, Siddiqui M, et al. Waterhouse-Friderichsen syndrome and bilateral renal cortical necrosis in meningococcal sepsis. Am J Kidney Dis 2000;36:396-400.
86. Hatherill M, Tibby SM, Hilliard T, et al. Adrenal insufficiency in septic shock. Arch Dis Child 1999;80:51-55.
87. Hodgetts TJ, Brett A, Castle N. The early management of meningococcal disease. J Accid Emerg Med 1998;15:72-76.
88. Riordan FA, Thomson AP, Sills JA, et al. Who spots the spots? Diagnosis and treatment of early meningococcal disease in children. BMJ 1996;313:1255-1256.
89. Inkelis SH, O'Leary D, Wang VJ, et al. Extremity pain and refusal to walk in children with invasive meningococcal disease. Pediatrics 2002;110(1 Pt 1):e3.
90. Lowenstein R. Deadly viral syndrome mimics. Emerg Med Clin North Am 2004;22:1051-1065, ix-x.
91. Hillman KM, Bristow PJ, Chey T, et al. Antecedents to hospital deaths. Intern Med J 2001;31:343-348.
92. Balmer P, Miller E. Meningococcal disease: How to prevent and how to manage. Curr Opin Infect Dis 2002;15:275-281.
93. Louria DB, Sen P, Kapila R, et al. Anterior thigh pain or tenderness. A diagnostically useful manifestation of bacteremia. Arch Intern Med 1985;145:657-658.
94. Granier S, Owen P, Pill R, et al. Recognising meningococcal disease in primary care: Qualitative study of how general practitioners process clinical and contextual information. BMJ 1998;316:276-279.
95. van de Beek D, de Gans J, Spanjaard L, et al. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med 2004;351:1849-1859.
96. Roos KL. Acute bacterial meningitis. Semin Neurol 2000;20:293-306.
97. Griffiss JM, Yamasaki R, Estabrook M, et al. Meningococcal molecular mimicry and the search for an ideal vaccine. Trans R Soc Trop Med Hyg 1991;85 Suppl 1:32-36.
98. Anderson J, Lind I. Characterization of Neisseria meningitidis isolates and clinical features of meningococcal conjunctivitis in ten patients. Eur J Clin Microbiol Infect Dis 1994;13:388-393.
99. Maini M, French P, Prince M, et al. Urethritis due to Neisseria meningitidis in a London genitourinary medicine clinic population. Int J STD AIDS 1992;3:423-425.
100. Vienne P, Ducos-Galand M, Guiyoule A, et al. The role of particular strains of Neisseria meningitidis in meningococcal arthritis, pericarditis, and pneumonia. Clin Infect Dis 2003;37:1639-1642.
101. Farron F, Cheseaux JJ, Pelet B. [Chronic meningococcemia and IgA deficiency in an adolescent]. Arch Pediatr 1996;3:149-151.
Neisseria meningitidis has the dubious distinction of being the last remaining serious bacterial threat to the lives and well being of otherwise healthy Americans. In industrialized nations, the risk of death or serious illness from organisms such as Haemophilus influenzae and Streptococcus pneumoniae has been reduced greatly by the development of vaccines against these encapsulated organisms.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.