Acetaminophen Toxicity
Acetaminophen Toxicity
Authors:
Michael Levine, MD, Department of Medical Toxicology, Banner Good Samaritan Medical Center, Phoenix, AZ.
Frank LoVecchio, DO, MPH, FACEP, ABMT, Department of Medical Toxicology, Banner Good Samaritan Medical Center, Phoenix, AZ; Department of Emergency Medicine Maricopa Medical Center, Phoenix, AZ.
Peer Reviewer:
Eric Lavonas, MD, FACEP, FACMT, Associate Director, Rocky Mountain Poison and Drug Center, Denver Health Emergency Physician, Denver Health Medical Center; Assistant Professor, Emergency Medicine, University of Colorado – Denver School of Medicine.
This article originally appeared in the November 23, 2009 issue of Emergency Medicine Reports. It was updated by Dr. Greg Wise in January 2011 with a revised editor's note and executive summary.
In June 2009, the advisors of the U.S. Food and Drug Administration recommended lowering the maximum dose of over-the counter acetaminophen, which is the key ingredient in such popular products such as Tylenol and Excedrin. The advisors were concerned that severe liver damage and even death can result from excessive ingestion of acetaminophen, which many consumers consider to be easier on the stomach than other medications and safe. The advisors recommended that the maximum daily dose of acetaminophen should be no more than 4 gm daily. The public is not thought to be sufficiently aware of the dangers of acetaminophen. Each year there are 56,000 emergency-room visits, 26,000 hospitalizations, and 458 deaths resulting from acetaminophen overdoses. Acetaminophen is the leading cause of acute liver failure in the United States, causing some 1,600 cases per year. The advisors' report, however, was not binding.
Now in January 2011, the FDA took action affecting only prescription drugs, not over-the-counter (OTC) medications. The FDA is requiring that within three years prescription pain drugs can contain no more than 325 mg of acetaminophen per pill or spoonful. Currently, some of these drugs contain as much as 750 mg of acetaminophen.
Prescription pain drugs will carry the FDA's strongest "black box" warning label. That label will warn of the risk of serious liver injury.
Nearly all the prescription drugs affected by the FDA action combine acetaminophen with an opioid. Popular brand names include Vicodin, Percocet, Lortab, Fioricet, and Roxicet. This issue reviews the consequences of acetaminophen toxicity. In light of these recommendations, primary care physicians need to better educate their patients regarding the dangers of acetaminophen and the need to be especially attentive to OTC drugs containing acetaminophen. The FDA's safety announcement can be found at the following URL: http://www.fda.gov/Drugs/DrugSafety/ucm239821.htm.
The Editor
Epidemiology
Acetaminophen, also known as paracetamol outside of the United States, was first discovered in the 1890s. Nearly 70 years passed before acetaminophen was first marketed as an analgesic or antipyretic. It was not until 1966 that Davidson described two cases of hepatotoxicity following acetaminophen ingestion.1 That same year, Thomason and colleagues also described a case of hepatotoxicity after paracetamol overdose.2 In the subsequent decades, as acetaminophen usage and overdoses became more prevalent, its potential to cause hepatotoxicity became well known. Today, acetaminophen is one of the most commonly used drugs in the treatment of pain or fever.3 Acetaminophen is available in a liquid preparation as well as in 325 mg, 500 mg, and 650 mg formulations. It is also combined with other drugs in nearly 200 different preparations.4 Frequently, acetaminophen is combined with codeine, propoxyphene (darvocet), hydrocodone (vicodin), and oxycodone (Percocet).
Much of the increased popularity of acetaminophen can be traced back to the desire to find a safer analgesic after salicylates were linked with Reye's syndrome in the 1980s.5 Since the 1980s, the use of acetaminophen and acetaminophen-containing products has increased substantially. In 2007, U.S. Poison Control Centers received more than 94,000 calls involving acetaminophen. Of these patients, acetaminophen was believed to be responsible for 348 deaths.6 Furthermore, nearly half of all cases of acute liver failure in the United States are believed to be due to acetaminophen.5
In recent months, there has been increased attention on the potential for hepatotoxicity following acetaminophen overdose. As a result, an advisory committee to the U.S. Food and Drug Administration (FDA) has recently begun hearings concerning acetaminophen and acetaminophen-induced hepatotoxicity.7 This concern, however, is not based on new data. The Medical Letter on Drugs and Therapeutics originally conducted an extensive review on acetaminophen toxicity in 2002. In light of the FDA's hearings, the same group re-reviewed the literature and concluded there were no more new data available in 2009 than there were during the original 2002 review.8
Pharmacokinetics/Pharmacodynamics
Following ingestion, acetaminophen is rapidly absorbed from the gastrointestinal tract. It has a relatively low volume of distribution (0.8-1.0 L/kg) and is 25% protein-bound.4 The half-life of acetaminophen is approximately 1.5-2.5 hours, although it can be slightly prolonged at supratherapeutic concentrations.9
Following ingestion, approximately 4% of the ingested acetaminophen is excreted unchanged in the urine, while the remainder is metabolized in the liver.10-11 In adults, 45-55% of acetaminophen is glucuronidated, while 20-30% is sulfonated.4 In pediatric patients, however, sulfation is the primary pathway, and glucuronidation is a minor component.12 The remainder of acetaminophen is metabolized via the cytochrome P450 isoenzyme CYP2E1 to form a substance called N-acetyl-para-benzoquinoneimine (NAPQI). Normally, the body's endogenous glutathione supplies are able to bind to and reduce NAPQI, resulting in renal excretion in the form of cysteine or mercaptopuric acid conjugates.11,13 In the setting of overdose, however, the NAPQI production exceeds the body's endogenous glutathione supply, and hepatotoxicity can result.4,11
Recently, research has demonstrated acetaminophen as a mitochondrial toxin. There are emerging data that NAPQI results in post-translational modifications of selected proteins involved in cellular and mitochondrial regulation and respiration.14 Ultimately a change in the membrane permeability occurs, resulting in impaired ATP synthesis and extrusion of mitochondrial proteins into the cellular cytoplasm. Cellular demise ultimately occurs.15
Histology of Liver Failure
Similar to several other toxins, acetaminophen can result in a relatively distinctive histologic pattern: centrilobular necrosis with passive congestion with scattered leukocytes without fatty infiltration.16 These areas of necrosis are characteristically followed by rapid disappearance of necrotic cells, which results in reticulin collapse. Among those who survive, there is a marked histologic recovery.17 Central lobular (zone three) necrosis can be seen with a number of toxic insults, including poisoning from Amaninta mushrooms, carbon tetrachloride, and copper, as well as with shock liver.
Risk Factors for Liver Failure
As previously stated, acetaminophen's toxicity is due to the metabolism to NAPQI by the cytochrome P450 isoenzyme CYP2E1. Thus, any xenobiotic that induces the P450 isoenzyme CYP2E1 can theoretically increase the risk for acetaminophen-induced hepatotoxicity. Perhaps one of the best-studied agents for inducing CYP2E1 is ethanol. Thus, chronic ethanol consumption leads to increased CYP2E1 activity and, in theory, subsequently increases the risk of hepatotoxicity (see Special Populations below).18 In contrast, the co-ingestion of ethanol with acetaminophen may result in inhibition of the microsomal oxidation of acetaminophen, thereby providing some degree of protection from acetaminophen-induced hepatotoxicity during acute intoxication.19 The strongest risk factor for developing hepatotoxicity, however, is the time from a toxic ingestion until the antidote, N-acetylcysteine, is administered. The risk of hepatotoxicity if N-acetylcysteine is started within the first eight hours of the overdose is exceedingly low, while the risk increases substantially with delays longer than eight hours.10
Clinical Presentation
Acute acetaminophen toxicity traditionally has been divided into four stages, with the time of each stage being approximate. The first stage, which occurs during the first 24 hours of overdose, primarily results in gastrointestinal symptoms. Nausea, vomiting, and anorexia are common. Right upper quadrant pain can begin toward the end of this stage. Patients can be asymptomatic, however, for much of this first stage. Because acetaminophen poisoning is common, and because early poisoning can be occult, most experts recommend obtaining acetaminophen levels on almost all acute intentional ingestions. The patient can remain asymptomatic or at least relatively asymptomatic in this stage; therefore, a high index of suspicion is needed to diagnose acetaminophen toxicity in a patient who is not admitting to an acetaminophen overdose. The second stage, which occurs from 24-72 hours post-ingestion, is characterized by the initial development of hepatic failure. While the vomiting and anorexia that are commonly encountered in the first stage can improve, right upper quadrant pain and tenderness frequently develop. Elevations of liver transaminases (AST and ALT) are common, and severe cases may develop metabolic acidosis (elevated lactate) and synthetic dysfunction (prolongation of the prothrombin time) at this stage. During the third phase, which can begin as early as 72 hours post-ingestion, the clinical symptoms depend on the degree of liver failure. Patients can remain relatively asymptomatic or can develop fulminant hepatic failure. Coma, encephalopathy, renal failure, coagulopathy, severe metabolic acidosis, and hypoglycemia can occur.10,15 At this stage, patients will either improve or will continue to worsen until they either die or receive a liver transplant. Stage four is characterized by resolution of liver damage. Thus, following stage III, the patient will either die or will have a complete recovery (stage IV). Unlike other causes of hepatic injury (e.g., alcohol-induced cirrhosis), acetaminophen-induced liver injury is not associated with long-term liver damage, assuming recovery occurs.
 |
Typically, the transaminitis is markedly elevated, and the aspirate aminotransferase (AST) can often exceed 10,000 IU/L. Frequently, one can observe a rise in the AST before the ALT. Metabolic acidosis and coagulopathy are common. Thrombocytopenia can occur,20 although its clinical significance is unclear. Hypophosphatemia can occur, and the degree of hypophosphatemia is directly correlated with the severity of the overdose.21 The degree of transaminases elevation does not predict outcome, but metabolic acidosis, synthetic dysfunction, renal insufficiency, and hepatic encephalopathy are independent predictors of death without transplant.22
Uncommonly, a significant anion-gap metabolic acidosis can occur early following massive acetaminophen ingestion. This early acidosis may result from the production and accumulation of 5-oxoproline (pyroglutamic acid).23 This acidosis is distinct from the acidosis that can develop late as part of fulminant hepatic failure, in which accumulating lactic acid is the primary etiology. Other uncommon features of acetaminophen poisoning are the development of pancreatitis or renal failure. While renal failure frequently can be encountered in the setting of hepatic failure, it can also occur, albeit uncommonly, in the absence of hepatic failure.24 Because the kidneys possess cytochrome P450, including the isoenzyme CYP2E1, it is possible that NAPQI production in the kidney causes direct renal injury.25 In treating a patient with severe acidosis following acetaminophen toxicity, it is prudent to rule out other potentially deadly treatable causes of anion-gap metabolic acidosis, such as salicylate poisoning, toxic alcohols, metformin ingestion, or shock, before attributing accumulation of 5-oxoproline as the cause.
Diagnosis and Treatment
The diagnosis of acetaminophen toxicity relies on history and laboratory studies. Because of vague symptoms in the early stage of acetaminophen toxicity, along with patients' occasional inadvertent confusion of acetaminophen, ibuprofen, and salicylates, it is recommended that all known or suspected overdoses have an acetaminophen level drawn. In addition, when one encounters a new diagnosis of hepatitis, a thorough medication history should be obtained. Because acetaminophen is present in so many different pharmaceutical agents, a patient may inadvertently overdose on acetaminophen by consuming multiple acetaminophen-containing products, each at the recommended dosage.
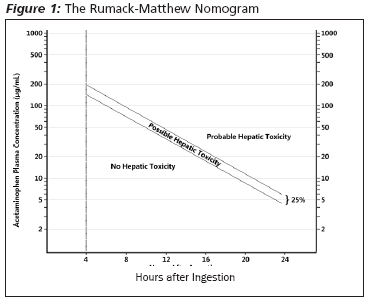
The need for treatment depends on the history, the physical examination, and laboratory studies. If treatment is desired, the preferred antidote is N-acetylcysteine, which was first proposed as a treatment for acetaminophen toxicity in 1974.26 An original paracetamol nomogram for acute poisoning was developed in Edinburgh, Scotland, in which a line on a semi-logarithmic graph joined two points: 200 mcg/dL at four hours and 30 mcg/dL at 15 hours.27 In the US, in order to improve sensitivity, the line was reduced by 25%.28 This revised nomogram uses a threshold of 150 mcg/mL at four hours after ingestion, and 75 mcg/mL eight hours after ingestion to determine the treatment line. Therefore, any detectable acetaminophen 24 hours or more after ingestion is an indication for treatment. This revised nomogram, which is the standard used in the United States, is referred to as the Rumack-Matthew nomogram. (See Figure 1.) Treatment with N-acetylcysteine is indicated when the acetaminophen concentration is above the line on the Rumack-Matthew nomogram.
 |
As with nearly any ingestion, the topic of gastrointestinal decontamination remains controversial. There is no role for the administration of syrup of ipecac in the emergency department management of acetaminophen ingestions or any overdose.10 Similarly, based on the risks and benefits, gastric lavage should not be used in the management of acetaminophen ingestions.29 There has been concern over the co-administration of activated charcoal with N-acetylcysteine. Specifically, some have raised concerns that the N-acetylcysteine will be absorbed by the charcoal, thus yielding sub-optimal concentrations of N-acetylcysteine. Spiller et al. examined this in a non-randomized trial and found no worse outcomes in those who received charcoal.30 Most likely, while the co-administration of charcoal with oral N-acetylcysteine does result in some decrease in the area under the curve of N-acetylcysteine, it is unlikely to be of any clinical consequence.10
N-acetylcysteine, the antidote of choice for acetaminophen poisoning, acts as a free radical scavenger as well as a gulathione substrate.31 Historically, N-acetylcysteine has been available in the United States only as an oral preparation, which is marketed under the name Mucomyst. While many different treatment durations have been proposed, the FDA-approved dosing regimen involves 140 mg/kg, followed by 70 mg/kg every four hours for 17 doses. This regimen requires 72 hours to complete. More recently, many other dosing strategies have been developed that involve less duration of therapy, and hence shorter hospital stays. In an observational study using oral N-acetylcysteine, Woo and colleagues demonstrated that a 36-hour course can be safe and effective.32 Similarly, Betten and colleagues demonstrated a 20-48 hour course of oral N-acetylcysteine can be safe and effective, although 4% of the patients had persistent abdominal pain after discharge with the shorter regimen.33 However, it should be noted that none of the patients in their study required readmission for these symptoms. Because both acetaminophen poisoning and oral N-acetylcysteine administration can cause nausea and vomiting, the administration of an oral antidote can be problematic in this setting.
 |
In 2004, the FDA approved Acetadote™, an intravenous N-acetylcysteine preparation. The intravenous route provides a safe and highly effective means to administer the antidote34-35 and has been the standard treatment modality in Europe for more than 30 years. The major adverse effect associated with the intravenous therapy, however, is the development of anaphylactoid reactions. Thus, the current FDA-approved regimen involves 150 mg/kg intravenously over 1 hour, followed by 12.5 mg/kg/hr for 4 hours, followed by 6.25 mg/kg/hr for 16 hours. While the anaphylactoid reactions are usually mild, at least one report of death in a "brittle asthmatic," as the authors describe, has resulted from the intravenous administration of N-acetylcysteine.36 Overall, however, anaphylactoid reactions typically are minor and can be treated easily with temporary discontinuation of the drug, along with the administration of diphenhydramine. Following the administration of diphenhydramine and improvement of symptoms, the infusion of N-acetylcysteine usually can be restarted without significant difficulty.37 Interestingly, the incidence of anaphylactoid reactions appears to be lower with high concentrations of acetaminophen.38-40 Prior to the availability of an intravenous N-acetylcysteine preparation in the United States (Acetadote™), patients who could not tolerate oral N-acetylcysteine were commonly treated by intravenous infusion of the oral preparation, filtered through a 0.22 micron filter.41
As a general rule, once the decision to treat has been made, repeat acetaminophen levels are unnecessary, except in cases of exceedingly high initial concentrations. Although the FDA-approved dosing regimens are of fixed durations, virtually all experts recommend tailoring the duration of therapy to the patient's clinical condition. After the patient has received at least 21-24 hours of therapy, transaminases and prothrombin level should be rechecked before discontinuing therapy. If the liver functions and prothrombin time are normal, and the patient is clinically well (neither vomiting nor complaining of abdominal pain), the N-acetylcysteine can be discontinued. Some institutions also recommend checking a repeat acetaminophen level before discontinuing therapy, although that is neither our practice nor an evidence-based practice.
It is important to recall that the Rumack-Matthew nomogram was designed for a single acute ingestion when the phlebotomy occurs between four and 24 hours post-ingestion. Many times patients do not present following a single acute ingestion, but rather consume some pills and a few hours later consume additional pills. In this case, in order to use the nomogram, the safest method is to assume all pills were ingested at the first time, and plot the level based on that time. For example, if a patient ingested 10 tablets of acetaminophen at noon and an additional 10 tablets at 1:00, an acetaminophen level should be obtained at 5:00, and treatment should be started if the concentration is above the line on the nomogram.
Not uncommonly, patients will present with an unknown time of ingestion. Occasionally this is because the patient is unwilling to tell, but more often this occurs in mixed ingestions in which the patient is encephalopathic or intubated and therefore not able to provide much history. In these cases, an acetaminophen level, liver function tests, and a prothrombin time (PT) should be obtained. If there is no detectable acetaminophen, and the liver functions and PT are normal, no treatment for the acetaminophen ingestion is needed. If there is any detectable acetaminophen, or if there are elevated transaminases or PT, treatment with N-acetylcysteine is indicated. Chronic ingestions in which the patients have taken supratherapeutic ingestions for several days should be managed the same as when the time of ingestion is not known.
As previously stated, the risk of hepatotoxicity increases when N-acetylcysteine is started more than eight hours post-ingestion. Thus, if a patient presents to the emergency department shortly after ingestion, N-acetylcysteine does not need to be started until an acetaminophen concentration can be plotted on the Rumack-Matthew nomogram. For example, if a patient presents two hours following an acetaminophen ingestion, N-acetylcysteine does not need to be started immediately but, rather, it is reasonable to wait for the four-hour concentration. If the patient presents close to eight hours, however, and an acetaminophen concentration cannot be obtained before the 8-hour time mark, one should consider empiric administration of N-acetylcysteine while waiting for the acetaminophen concentration to return.
Because of its low volume of distribution and minimal protein binding, acetaminophen can be removed via hemodialysis. Because N-acetylcysteine is so effective in the management of acetaminophen toxicity, the role for extracorporeal removal is minimal. However, it should be noted that in cases of massive acetaminophen ingestions, extracorporeal elimination can be considered,42 especially if there is a co-existing metabolic acidosis.
In 1994, McNeil Pharmaceuticals released an "extended-release formulation" of acetaminophen marketed under the name Tylenol Extended Relief™. This bi-layered capsule contained 325mg of acetaminophen that was immediately available, and an additional 325 mg that was available after the bilayer capsule was penetrated. However, because the pharmacokinetic profile between the regular release and the extended release formulation are similar, it appears both safe and reasonable to use a single four-hour acetaminophen concentration and determine treatment based on the Rumack-Matthew nomogram for either the regular release or the extended release acetaminophen products.10
Differential Diagnosis
While the diagnosis of acetaminophen-induced liver failure may be obvious in a patient who presents with a suicide note and an empty bottle of acetaminophen, frequently the emergency physician is confronted with a patient in liver failure with no history of overdose. In these cases, the acetaminophen concentration is often negative, as it has been metabolized completely by the time hepatic failure occurs. Nonetheless, it is important to consider acetaminophen on the differential diagnosis of any patient with hepatic failure.
While many drugs can cause a toxin-induced hepatitis, fewer xenobiotics can cause fulminant hepatic failure. Among these drugs are acetaminophen, amoxicillin-clavulanate, isoniazid, nitrofurantoin, iron, herbals (e.g. pennyroyal and chaparral), non-steroidal anti-inflammatory medications (especially diclofenac or sulindac), phenytoin, and the thiazolidinedione troglitazone. In addition, the recreational drugs cocaine and 3,4-methylenedioxymethamphetamine (MDMA or ecstasy), as well as carbon tetrachloride, white phosphorus, arsenic, thallium, and borates can cause acute hepatic failure. It should be noted that the cause of acute hepatic failure is dose-dependent in some of these agents (e.g. acetaminophen), yet is idiosyncratic with other agents (e.g. troglitazone). Furthermore, any drug that produces profound hypotension can result in hepatic failure from shock liver. In addition, many drugs, such as the HMG-CoA reductase inhibitors ("statins") can cause a liver injury, but not fulminant hepatic failure.
While a full review of all causes of hepatic failure is beyond the scope of this paper, the emergency physician should always consider infectious etiologies (e.g., hepatitis B, hepatitis C, Epstein Barr virus [EBV], herpes simplex virus [HSV], and cytomegalovirus [CMV]). Other causes include vascular etiologies (e.g., Budd-Chiari syndrome, ischemic hepatitis, veno-occlusive disorders), and miscellaneous etiologies such as Wilson's disease, autoimmune hepatitis, fatty liver of pregnancy, heat stroke, and HELLP syndrome.
While the history may be helpful in distinguishing the etiology of fulminant hepatic failure, several additional laboratory features might be helpful as well. Frequently, the asparate aminotransferase (AST) rises faster and peaks sooner than the alanine aminotransferase (ALT). In addition, unlike some other etiologies of hepatic failure, there is synthetic dysfunction occurring as well, which results in a rise in the prothrombin time.
Liver Failure
The greatest concern following acetaminophen ingestions is the potential to develop hepatotoxicity. The risk of hepatotoxicity is substantially increased when N-acetylcysteine therapy is begun more than 8-10 hours post-ingestion.43 The development of liver failure is characterized not only by the development of transaminitis, but also encephalopathy, acidosis, renal failure, coagulopathy, and hypoglycemia. The proximate cause of death in acetaminophen-induced hepatic failure is usually cerebral edema.
N-acetylcysteine should be continued until one of three major endpoints happens: clinical and laboratory improvement; the patient receives a liver transplant; or death. The clinical improvement is primarily characterized as resolution of encephalopathy, while the laboratory improvement includes improvement in the alanine aminotransferase, prothrombin time, and creatinine.13 While one recent study in mice has questioned the benefit of continued therapy with N-acetylcysteine,44 the use of prolonged acetylcysteine dosing is supported by a randomized human clinical trial.45
Raschke and colleagues recently published a protocol for management of patients in fulminant liver failure. In their series of 22 patients with fulminant liver failure and a grade three or four encephalopathy, 12/22 (55%) were due to acetaminophen. In addition to acetylcysteine, their protocol involved placement of an intracranial pressure monitor, aggressive management of intracranial hypertension, and vasopressors titration. This strategy was associated with good clinical outcomes among the 18 transplant candidates, including those who did not ultimately receive a liver transplant.46 Importantly, none of the patients died from cerebral edema, which is the most common cause of death in acetaminophen overdose.
Special Populations
Pregnancy. Acetaminophen readily crosses the placenta, which places the fetus at potential risk of hepatotoxicity.11,47 However, because NAPQI does not cross the placenta, the fetus itself must metabolize the acetaminophen in order for hepatotoxicity to occur. The fetus is able to start metabolizing acetaminophen into both toxic and non-toxic metabolites beginning at approximately 18 weeks gestational age.11 N-acetylcysteine does cross the placenta in both animal models48 and humans,49 and its use is indicated in pregnant women whose serum concentration is above the treatment line on the Rumak-Matthew nomogram. Fetal outcome appears to be worse with delays in commencing N-acetylcysteine.50
Alcoholics. Chronic alcohol consumption results in depletion of hepatic glutathione and upregulation of CYP2E1,18 the isoenzyme which metabolizes acetaminophen to NAPQI. As such, there is concern that chronic alcoholics may be more susceptible to acetaminophen-induced hepatotoxicity. Kuffner and colleagues examined the effects of administering 4 grams of acetaminophen daily to alcoholics entering an alcohol detox facility. These patients are hypothesized to be at the highest risk of hepatotoxicity, as at the time these patients enter a detox facility, they likely have their lowest glutathione supplies while being at maximal induction of CYP2E1. While at the maximal recommended daily dose, they failed to demonstrate any increased risk of hepatotoxicity, including in those patients with alcoholic hepatitis.51-52 However, chronic alcohol abuse is associated with worsened outcomes in acute acetaminophen poisoning.53
Opiates. Watkins and colleagues attempted to determine if the co-administration of opiates along with acetaminophen resulted in increased hepatotoxicity. In their study, 147 patients were randomized to receive either placebo, acetaminophen, morphine and acetaminophen, hydromorphone and acetaminophen, or oxycodone and acetaminophen. All patients who received acetaminophen received 4 grams daily. Among the placebo, only 1 patient had a rise in the ALT twice the upper limit of normal. In contrast, more than 19% of participants in each of the four active treatment groups had an ALT five times the upper limit of normal. All three of the opioid/acetaminophen treatments frequently resulted in elevation of the ALT.54 Thus, the addition of opiates to acetaminophen at doses of 4 grams daily did not produce any increase in the incidence of subclinical hepatotoxicity.
Pediatrics. Pediatric patients appear to be somewhat resistant to acetaminophen-induced hepatotoxicity when compared with adults.55 It appears that the increased rate of sulfation along with possibly an increased relative size of the liver, affords some hepato-protective effects to pediatric patients. In fact, many authors have suggested that 200 mg/kg should be the potentially hepatic-toxic dose in pediatrics, as compared with the lower dose for adults.55-57
Prognosis
In general, patients who have N-acetylcysteine started within 8 hours of the overdose will do well. Once hepatic injury occurs, the King's College Criteria can be used to help predict which patients may benefit from liver transplant.58 According to these criteria, patients should be considered for liver transplant if 24 hours after admission the arterial pH is less than 7.25 after fluid resuscitation, or if there is a combination of a prothrombin time longer than 100 seconds, grade III or IV encephalopathy, and a serum creatinine higher than 3.4 mg/dL.58
Prevention
Because of the risk of hepatotoxicity following overdoses on acetaminophen, various strategies have been proposed to reduce the availability of the drug. One such strategy involved limiting the number of acetaminophen tablets that could be sold in an individual package. In 1988, the government of the United Kingdom (UK) reduced the number of tablets of acetaminophen that could be sold outside a pharmacy to 16 tablets or capsules. Several studies have examined the effectiveness of such a strategy and have found mixed results. Thus, it is not clear if reducing the number of tablets per box is associated with an overall reduction in toxicity.59
Pitfalls in Management
There are several common scenarios in which mistakes in management commonly occur. Perhaps the most common source of error is failure to interpret the acetaminophen level in the context of the time ingested. A single acetaminophen level is not helpful without knowing the time of ingestion. Thus, it is critical to interpret the acetaminophen level based on the time of ingestion and plot the concentration on the Rumack-Matthew nomogram. If the time of ingestion is not known, it is important that the patient be started on N-acetylcysteine if any acetaminophen is detected or if there are abnormal liver function tests or prothrombin time.
A second common source of error is discontinuing the N-acetylcysteine at 21 hours simply because the time course for therapy is over. If the patient is having rising liver function tests or persistent right upper quadrant tenderness, then therapy with N-acetylcysteine should be continued until these are improving. Patients meeting criteria for transplantation should be referred when indicated. A third common source of error for the emergency physician is failure to exclude other ingestions. Even if a patient admits to ingesting acetaminophen, other potentially life-threatening ingestions, including salicylates, need to be evaluated in all patients. Lastly, a common source of error for the emergency physician is failure to obtain appropriate psychiatry consult when indicated. If a patient is being admitted to the hospital, then the consultation can be done once the patient is admitted, but if the initial acetaminophen level does not warrant therapy and the patient is to be medically cleared from the emergency department, it is important to obtain a psychiatric consultation if indicated.
Summary
Acetaminophen overdose remains a common cause of hepatic failure. To ensure optimal care, the diagnosis must be made early, and treatment started within 8 hours. With prompt treatment with N-acetylcysteine, the incidence of hepatic failure can be reduced.
References
1. Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. BMJ 1966;2:497-499.
2. Thomson JS, Prescott LF. Liver damage and impaired glucose tolerance after paracetamol overdosage. BMJ 1966;2:506-507.
3. Kaufman DW, Kelly JP, Rosenberg L, et al. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA 2002;287:337-344.
4. Baselt RC. Disposition of Toxic Drugs and Chemicals in Man. 7th edition. Biomedical Publications. Foster City, CA. 2004; 6-9.
5. Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: Lowering the risks of hepatic failure. Hepatology 2004;40:6-9.
6. Bronstein AC, Spyker DA, Cantilena LR, et al. 2007 Annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 25th Annual Report. Clin Toxicol 2008;46:927-1057.
7. Department of Health and Human Services. Food and Drug Administration. Docket No. FDA-2009-N-0138. Federal Register 2009;74:18731.
8. Anonymous. Acetaminophen safety déjà vu. Med Lett Drugs Ther 2009;51:53-54.
9. Prescott LF. Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmac 1980;10:291s-298s
10. Zed PJ, Krenzelok EP. Treatment of acetaminophen overdose. Am J Health Syst Pharm 1999;56:1081-1091.
11. Wilkes JM, Clark LE, Herrera JL. Acetaminophen overdose in pregnancy. South Med J 2005;98:1118-1122.
12. Rumore MM, Blaiklock RG. Influence of age-dependent pharmacokinetics and metabolism on acetaminophen hepatotoxicity. J Pharm Sci 1992;81:203-207.
13. Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med 2008;359:285-292.
14. Andringa KK, Bajt ML, Jaeschke H, et al. Mitochondrial protein thiol modifications in acetaminophen hepatotoxicity: Effect on HMG-CoA synthase. Toxicol Lett 2008;177:188-197.
15. Chun LJ, Tong MJ, Busuttil RW, et al. Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol 2009;43:342-349.
16. Rowen AK, Norvell J, Elridge DL, et al. Acetaminophen poisoning. Clin Lab Med 2006;26:49-65
17. Portmann B, Talbot IC, Day DW, et al. Histopathological changes in the liver following a paracetamol overdose: Correlation with clinical and biochemical parameters. J Pathol 1975;117:169-181.
18. Riordan SM, Williams R. Alcohol exposure and paracetamol-induced hepatotoxicity. Addict Biol 2002;7:191-206.
19. Prescott LF. Paracetamol, alcohol, and the liver. Br J Clin Pharmacol 2000;49:291-301.
20. Fischereder M, Jaffe JP. Thrombocytopenia following acute acetaminophen overdose. Am J Hematol 1994;45:258-259.
21. Jones AF, Harvey JM, Vale JA. Hypophosphatemia and phosphaturia in paracetamol poisoning. Lancet 1989;334:608-609.
22. O'Grady JG, Alexander GJ, Hayllar KM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989;97:439-445.
23. Fenves AZ, Kirkpatrick HM 3rd, Patel VV, et al. Increased anion gap metabolic acidosis as a result of 5-oxoproline (pyroglutamic acid): A role for acetaminophen. Clin J Am Soc Nephrol 2006;1:441-447.
24. Jones AF, Vale JA. Paracetamol poisoning and the kidney. J Clin Pharm Ther 1993;18:5-8.
25. Mazer M, Perrone J. Acetaminophen-induced nephrotoxicity: Pathophysiology, clinical manifestations, and management. J Med Toxicol 2008;4:2-6.
26. Prescott LF, Matthew H. Cysteamine for paracetamol overdosage. Lancet 1974;1:998.
27. Prescott LF, Illingwort RN, Critchley JA, et. al. Intravenous N-acetylcystine: The treatment of choice for paracetamol poisoning. BMJ 1979;2:1097-1100.
28. Rumack BH, Peterson RC, Koch GG, et. al. Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch Intern Med 1981;141 (3 Spec No):380-385.
29. Vale JA, Kulig K. American Academy of Clinical Toxicology; European Association of Poison Centers and Clinical Toxicologist. Position paper: Gastric lavage. J Toxicol Clin Toxicol 2004;42:933-943.
30. Spiller HA, Krenzelok EP, Grande GA, et. al. A prospective evaluation of the effect of activated charcoal before oral N-acetylcysteine in acetaminophen overdose. Ann Emerg Med 1994;23:519-523.
31. Bruno MK, Cohen SD, Khairallah EA. Antidotal effectiveness of N-acetylcysteine in reversing acetaminophen-induced hepatotoxicity. Enhancement of the proteolysis of arylated proteins. Biochem Pharmacol 1988;37:4319-4325.
32. Woo OF, Mueller PD, Olson KR, et al. Shorter duration of oral N-acetylcysteine therapy for acute acetaminophen overdose. Ann Emerg Med 2000;35:363-368.
33. Betten DP, Cantrell FL, Thomas SC, et al. A prospective evaluation of shortened course oral N-acetylcysteine for the treatment of acute acetaminophen poisoning. Ann Emerg Med 2007;50:272-279.
34. Whyte IM, Francis B, Dawson AH. Safety and efficacy of intravenous N-acetylcysteine for acetaminophen overdose: Analysis of the Hunter Area Toxicology Service (HATS) database. Curr Med Res Opin 2007;23:2359-2368.
35. Whyte AJ, Kehrl T, Brooks DE, et al. Safety and effectiveness of acetadote for acetaminophen toxicity. J Emerg Med 2008 Nov 18 (Epub).
36. Appelboam AV, Dargan PI, Knighton J. Fatal anaphylactoid reaction to N-acetylcysteine: Caution in patients with asthma. Emerg Med J 2002;19:594-595.
37. Bailey B, McGuigan MA. Management of anaphylactoid reactions to intravenous N-acetylcysteine. Ann Emerg Med 1998;31:710-715.
38. Waring WS, Stephen AF, Robinson OD, et al. Lower incidence of anaphylactoid reactions to N-acetylcysteine in patients with high acetaminophen concentrations after overdose. Clin Toxicol (Phila) 2008;46:496-500.
39. Pakravan N, Waring WS, Sharma S, et al. Risk factors and mechanisms of anaphylactoid reactions to acetylcysteine in acetaminophen overdose. Clin Toxicol (Phila) 2008;46:697-702.
40. Lynch RM, Robertson R. Anaphylactoid reactions to intravenous N-acetylcysteine: A prospective case controlled study. Accid Emerg Nurs 2004;12:10-15.
41. Lavonas EJ, Beuhler MC, Ford MD, et al. Intravenous administration of N-acetylcysteine: Oral and parenteral formulations are both acceptable. Ann Emerg Med 2005;45:223-224.
42. Ash SR, Caldwell CA, Singer GG, et al. Treatment of acetaminophen-induced hepatitis and fulminant hepatic failure with extracorporeal sorbent-based devices. Adv Ren Replace Ther 2002;9:42-53.
43. Smilkstein MJ, Knapp GL, Kulig KW, et al. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988;319:1557-1562.
44. Yang R, Miki K, He X, et al. Prolonged treatment with N-acetylcysteine delays liver recovery from acetaminophen hepatotoxicity. Crit Care 2009;13:R55.
45. Harrison PM, Keays R, Bray GP, et al. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet 1990;335:1572-1573.
46. Rashke RA, Curry SC, Gerkin R, et al. Results of a protocol for the management of patients with fulminant liver failure. Crit Care Med 2008;36:2244-2248.
47. Wang LH, Rudolph AM, Benet LZ. Pharmacokinetic studies of the disposition of acetaminophen in the sheep maternal-placental fetal unit. J Pharmacol Exp Ther 1986;238:|198-205.
48. Selden BS, Curry SC, Clark RF, et al. Transplacental transport of N-acetylcysteine in an ovine model. Ann Emerg Med 1991;20:1069-1072.
49. Horowitz RS, Dart RC, Jarvie DR, et al. Placental transfer of N-acetylcysteine following human maternal acetaminophen toxicity. Toxicol Clin Toxicol 1997;35:447-451.
50. Riggs BS, Bronstein AC, Kulig K, et al. Acute acetaminophen overdose during pregnancy. Obstet Gynecol 1989;74:247-253.
51. Kuffner EK, Green JL, Bogdan GM, et al. The effect of acetaminophen (four grams a day for three consecutive days) on hepatic tests in alcoholic patients a multicenter randomized study. BMC Med 2007;5:14.
52. Kuffner EK, Dart RC, Bogdan GM, et al. Effect of maximal daily doses of acetaminophen on the liver of alcoholic patients: A randomized, double-blind, placebo-controlled trial. Arch Intern Med 2001;161:2247-2252.
53. Sivilotti ML, Yarema MC, Juurlink DN, et al. A risk quantification instrument for acute acetaminophen overdose patients treated with N-acetylcysteine. Ann Emerg Med 2005;46:263-271.
54. Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: A randomized controlled trial. JAMA 2006;296:87-93.
55. Burillo-Putze G, Mintegui S, Munne P. Changes in pediatric toxic dose of acetaminophen. Am J Emerg Med 2004;22:323.
56. Bond GR, Krenzzelok EP, Normann SA, et al. Acetaminophen ingestion in childhood-cost and relative risk of alternative referral strategies. J Toxicol Clin Toxicol 1994;32:513-525.
57. Moher CR, Nordt SP, Williams SR, et al. Prospective evaluation of mild to moderate pediatric acetaminophen exposures. Ann Emerg Med 2000;35:239-244.
58. Anand AC, nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: An assessment of the King's criteria. J Hepatol 1997;26:62-68.
59. Hawkins LC, Edwards JN, Dargan PI. Impact of restricting paracetamol pack sizes on paracetamol poisoning in the United Kingdom: A review of the literature. Drug Saf 2007;30:465-479.
In June 2009, the advisors of the U.S. Food and Drug Administration recommended lowering the maximum dose of over-the counter acetaminophen, which is the key ingredient in such popular products such as Tylenol and Excedrin. The advisors were concerned that severe liver damage and even death can result from excessive ingestion of acetaminophen, which many consumers consider to be easier on the stomach than other medications and safe.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.