Drug Interactions and the Medical Home
AUTHORS
Brian T. Hocum, PharmD, CGP, Adjunct Faculty Member, Washington State University College of Pharmacy, Spokane, WA; Regional Pharmacist Liaison, Genelex Corporation, Seattle, WA
Elizabeth L. Black, MD, Blue Mountain Family Health, Clarkston, WA
Kevin J. Black, MAT, Clarkston, WA
PEER REVIEWER
Stuart J. Beatty, PharmD, BCACP, CDE, Associate Professor of Clinical Pharmacy, The Ohio State University College of Pharmacy, Columbus, OH
EXECUTIVE SUMMARY
Management of pharmacotherapy is emerging as a principal task of primary care physicians. In their role as coordinators of patient care, effective monitoring of medication management is crucial to achieving the goals of the patient-centered medical home.
- Drug-to-drug interactions represent one of the most significant risk of polypharmacy in our increasingly complex and aging populations.
- Before the advent of useful software, physicians were remarkably inaccurate in recognizing and classifying interactions, even with strongly contraindicated medication pairings.
- A better understanding of the interactions related to drug absorption, distribution, metabolism, and excretion will have salutary benefits in patient management.
- The changing formulary requirements of insurance payers, the elusive balance of software systems to effectively warn of significant interactions without resulting in alert fatigue, the dizzying increase in drug choices and complexity, and the emergence of the patient-centered medical home with its accompanying expectations to create value and improved outcomes mandate the cooperative interaction between primary care physicians and clinical pharmacists.
INTRODUCTION
Drug-to-drug interactions (DDI) are a concern of increasing significance at all levels of health care. This article will provide case examples and illustrations to demonstrate the metabolic processes that underlie many of these interactions. An understanding of the fundamental mechanisms of drug metabolism and transport at the cellular level is increasingly important for primary care providers. Understanding these mechanisms can be especially useful in actively assimilating and even exploiting DDI information as part of patient care management.
Throughout the history of medicine, pharmaceuticals have often been deployed with an understanding of their effects, but a much poorer understanding of how those effects occur at the deeper metabolic level. Drug metabolism is a complex subject that is under active investigation by pharmaceutical science. The primary mechanisms that will be discussed in this article are transporter proteins that carry drug molecules into and out of cellular systems, where they undergo various phases of metabolism, and the enzyme families that are involved in the transformation of these drug molecules.
The mechanisms of drug metabolism have an interesting evolutionary history. The metabolic systems that are relied on to transmit drugs through the body were not developed to introduce beneficial substances into the body, but rather to keep harmful substances out. As animal ancestors diverged from plants and began relying on them as a food source, plants developed phytoalexins that could poison and deter the animals that fed on them. Through selective pressures, animals with more robust enzymatic systems that could neutralize those toxins thrived. This back and forth “animal and plant warfare” resulted in diversification of enzymes that now metabolize drugs in the body.1
As a better understanding of drug metabolism emerges, pharmacotherapy is increasingly able to adapt to this metabolic defense mechanism to introduce beneficial drugs into the body more effectively or more efficiently. Medications are increasingly being developed specifically to either avoid or exploit particular metabolic processes to achieve better efficacy or decrease the potential for interaction with other medications that use the same metabolic pathways. These metabolic points of interaction between drugs are the primary focus of this article.
Management of pharmacotherapy is emerging as one of the principal tasks of primary care providers (PCPs). In their role as coordinators of patient care, PCPs have always sought to provide effective care for their patients. Emerging models, including the patient-centered medical home (PCMH), place significant emphasis on this central role of the PCP as the coordinator of patient care. Measuring and monitoring patient outcomes and progress toward specific treatment goals play a significant role in the PCMH model. Increasingly, PCPs are also being asked by private and public insurance to demonstrate their effectiveness at achieving treatment goals within their patient populations by reporting on quality data.
The physician quality reporting system (PQRS) has numerous measures that seek information on treatment goals associated with specific diagnoses. Blood pressure, cholesterol levels, and A1C levels are becoming more than just an indicator for the physician of how well a medical condition is being managed. These values, taken in aggregate over the entire patient panel of a given provider, are being used by payers and other external entities to evaluate how well providers are managing their patient populations.
Although initially deployed as a voluntary system, PQRS has utilized both incentives and increasing penalties in the form of reductions to the Centers for Medicaid and Medicare Services (CMS) reimbursement to encourage providers to report on these kind of data. As of 2015, groups of 20 or more providers will have CMS compensation adjusted on the basis of how well their physicians perform on such data-driven measures of health care quality.
The ramifications of this trend toward data-driven definitions of successful treatment remain to be seen. Regardless, the pressure to demonstrate achievement of treatment goals in the form of objective data seems likely to increase. Providers will therefore have significant incentive to utilize pharmacotherapy to help patients meet the defined target goals quickly and effectively. As patients age and the number of comorbid conditions increases, more medications will be employed to meet multiple treatment goals. This will inevitably increase the risk of polypharmacy in these patients (see Figure 1).
Figure 1. Polypharmacy Comorbidities
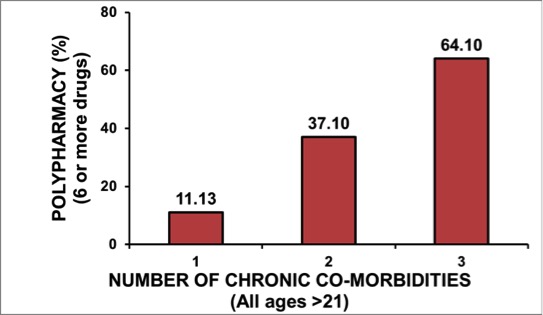
DDIs represent one of the most significant risks of polypharmacy. As the number of medications involved in a patient’s treatment regimen increases, the likelihood of any two of those medications having an interaction that significantly affects the action of one or both of the medications involved increases dramatically (see Figure 2). Providers with access to electronic medical records (EMRs) and electronic prescribing (ERx) software are becoming increasingly familiar with interaction alerts that are automatically generated by such systems to help anticipate DDIs. Before the widespread adoption of these software tools, health care providers were not ideally successful at anticipating or evaluating DDIs on their own. One study showed that a group of physicians was able to recognize or classify such interactions only just over half the time, even with strongly contraindicated medication pairings.2 Clearly, these software tools are a necessary adjunct to successful management of pharmacotherapy. As polypharmacy becomes more endemic, however, these alerts may become so frequent that providers may no longer be able to easily discern which alerts are most likely to dramatically impact the overall safety and efficacy of the medications involved. This is especially important, as alerts generated by these systems do not always represent DDI of sufficient clinical significance to avoid certain medication combinations. A more recent study showed that providers’ understanding of the relative clinical significance of DDI was “generally poor.”3 This further highlights the importance of improving provider understanding of the mechanisms underlying DDI.
Figure 2.
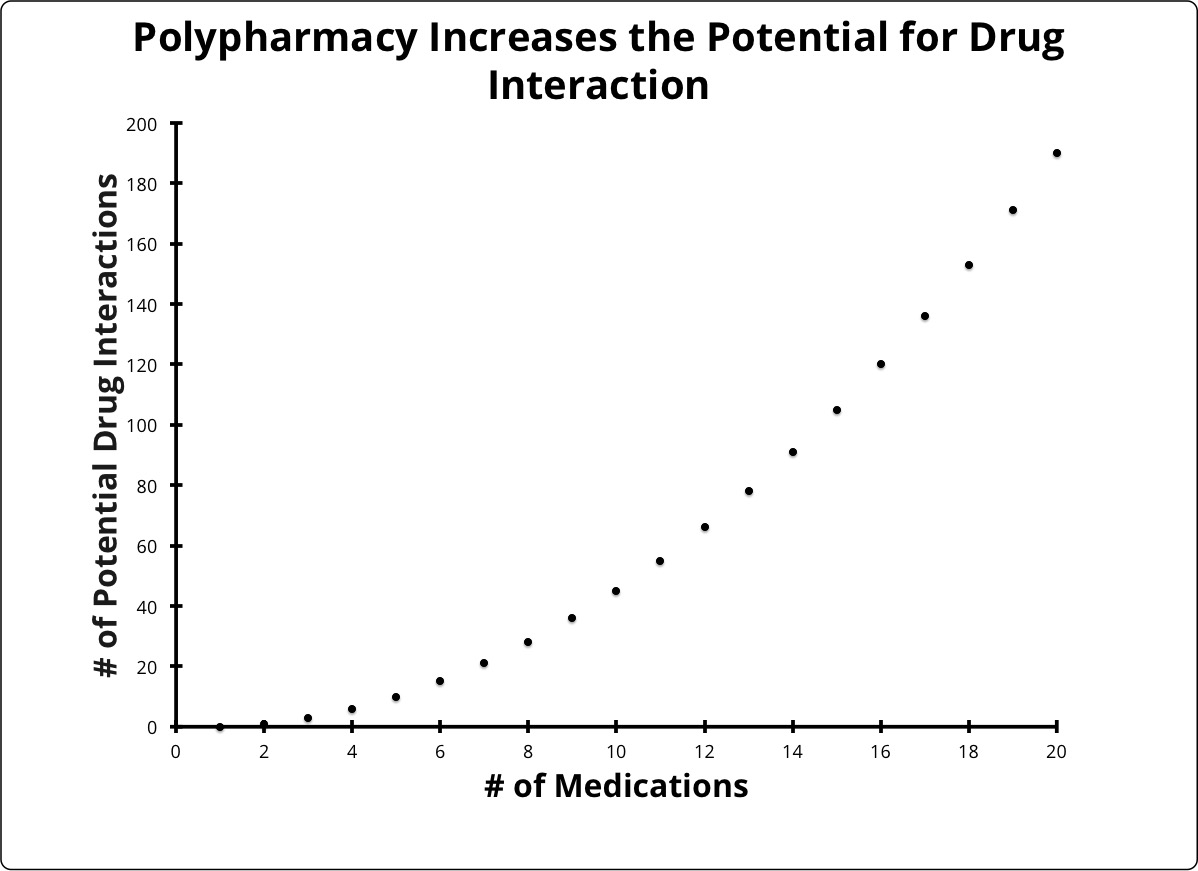
Guidance is even less clear in interactions that may affect the potential efficacy of a medication in the patient, but are not clearly contraindicated. Coadministration of a particular pair of medications may adversely affect the effectiveness of one or both of the medications, but not to the point where they are entirely ineffective in achieving desired goals. Alternatively, some drug combinations may actually reinforce the effectiveness of one medication to the point that they can be used at lower doses than are usually necessary to achieve the desired therapeutic results.
The above interactions illustrate how drug absorption, distribution, metabolism, and excretion (ADME) can affect the action of pharmaceutical agents. It is our hope that as a result of this discussion, PCPs may be encouraged and empowered to further explore the subject. This deeper understanding will better position PCPs to monitor for key indicators of harm associated with DDIs, including adverse drug reactions, toxicity, or treatment failures stemming from impaired medication effectiveness. The ability to monitor for and recognize these indicators as potential symptoms of unanticipated DDI is also beneficial, since this knowledge will facilitate a more rapid recognition of DDIs when they do occur. Although an understanding of pharmacokinetics clearly enhances the PCP’s ability to interpret and anticipate DDI in the context of polypharmacy, it may also be used to the PCP’s advantage in achieving desired therapeutic goals.
Absorption
An excellent place to begin the discussion of drug interactions is with the absorption of orally administered medications. Before oral medications can reach systemic circulation and their site of action, they must be absorbed. Multiple factors, including gastric pH and motility, gut pathology, and the chemical structure and formulation of the medication itself, can influence this.4 These factors can be manipulated to achieve increased or decreased blood levels of drugs and desired therapeutic outcomes. For example, the presence or absence of food may dramatically influence gastrointestinal (GI) motility.
GI motility is more rapid on an empty stomach.5 Some medications, like diazepam, when taken on an empty stomach, reach the small intestine more rapidly, where they are absorbed into the bloodstream (see Table 1-1). This may result in a faster time to reach the maximum concentration (Cmax) and, thus, maximum effectiveness, compared to taking the same medications with food. The presence of food tends to slow gastric motility. Although this may delay the time it takes for the drug to reach Cmax, the increased time in the gut may allow more time for the medication to enter into solution, which is required for absorption. Thus, the overall exposure to the drug, as measured by the area under the curve (AUC), may be greater. Composition of foods can also alter the absorption of drugs. Foods rich in fat, fiber, or calcium have a significant effect on the absorption of many medications.
Table 1. Gastrointestinal/Absorption Interaction Examples
|
Drug Combination |
Mechanism |
Effect |
|
|
1. Diazepam [package insert]. Teva Pharmaceuticals, Sellersville, PA; January 2014. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=48aa32cb-047a-414a-822e-82a5f26d881 Accessed December 28, 2014. 2. Metformin [package insert]. Solco Healthcare, Cranbury, NJ; January 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=bfdc6d04-2859-4a0c-add9-81ab131526ca Accessed December 28, 2014. 3. Amphetamine Mixed Salts [package insert]. Mallinckrodt Inc., Hazelwood, MO; November 2014. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=72ddd1c9-ddbd-4c95-acd9-003189a353a3 Accessed December 28, 2014. 4. Singh N, Singh PN, Hershman JM. Effect of calcium carbonate on the absorption of levothyroxine. JAMA 2000;283:2822-2825. 5. Atorvastatin [package insert]. Apotex Corp., Weston, FL; June 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=d9adb9e4-c495-9530-e5e3-7e3b01d53e4c Accessed December 29, 2014. 6. Kantola T, Kivisto KT, Neuvonen PJ. Grapefruit juice greatly increases serum concentrations of lovastatin and lovastatin acid. Clin Pharmacol Ther 1998;63:397-402. 7. Simvastatin [product insert]. Cobalt Laboratories, Bonita Springs, FL; February 2010. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b3022011-4c11-7516-1f6e-ef606f5954a4 Accessed December 29, 2014. 8. Smith CL, Hampton EM, Pederson JA, et al. Clinical and medico-economic impact of the cyclosporine-diltiazem interaction in renal transplant recipients. Pharmacother 1994;14:471-481. 9. Kilby JM, Sfakianos G, Gizzi N, et al. Safety and pharmacokinetics of once-daily regimens of soft-gel capsule saquinavir plus minidose ritonavir in human immunodeficiency virus-negative adults. Antimicrob Agents Chemother 2000;44:2672-2678. |
|||
|
1 |
diazepam + food |
food interaction |
Diazepam and a moderate fat meal resulted in a 27% decrease to diazepam AUC, a 20% decrease in Cmax and a 30-minute delay in absorption (15 minutes vs 45 minutes).1 |
|
2 |
metformin + food |
food interaction |
Single-dosed metformin 500 mg to 2500 mg with either a low-fat or high-fat meal resulted in a 38% or 73% increase to metformin AUC, respectively, relative to fasting.2 |
|
3 |
amphetamines + GI acidifiers |
pH interaction |
GI acidifiers (e.g. non-diet soda, ascorbic acid, cranberry and other fruit juices) increase ionization of amphetamines and lower absorption of amphetamines.3 |
|
4 |
amphetamines + GI alkalinizers |
pH interaction |
GI alkalinizers decrease ionization of amphetamines and increase absorption of amphetamines.3 |
|
5 |
levothyroxine + calcium carbonate |
formation of insoluble complex |
Multidosed calcium carbonate 1200 mg daily added to a previously stabilized dose of levothyroxine x 3 months resulted in a 9% decrease in mean free T4 and a 7% decrease in mean total T4 levels. 20% of patients had serum TSH levels higher than the normal range during calcium carbonate coadministration.4 |
|
6 |
statins + grapefruit |
CYP3A4 inhibition |
Single-dosed atorvastatin 40 mg and multidosed grapefruit juice 240 mL daily resulted in a 37% increase in atorvastatin AUC.5 Multidosed grapefruit juice 200 mL double strength TID x 3 days and single-dosed lovastatin 80 mg resulted in a 1400% increase in lovastatin AUC and a 400% increase in metabolite lovastatin acid AUC.6 Multidosed grapefruit juice single-strength 8 ounces x 3 days and single-dosed simvastatin 20 mg on day 3 resulted in 90% increase in simvastatin AUC and 30% increase in metabolite simvastatin acid AUC.7 |
|
7 |
cyclosporine + diltiazem |
CYP3A4 & P-gp inhibition |
Multidosed diltiazem 90 mg to 120 mg PO BID and oral cyclosporine resulted in a 51% increase in cyclosporine AUC and a 34% increase in Cmax (1999 Asberg paper). In another study the dose of cyclosporine was decreased such that an estimated 28% cost savings per patient was achieved.8 |
|
8 |
protease inhibitors (e.g., saquinavir) + ritonavir |
CYP3A4 & P-gp inhibition |
Saquinavir and ritonavir resulted in up to a 592% increase in saquinavir AUC and 424% increase in Cmax.9 |
|
Tables may not include entire mechanisms or studies with conflicting findings. Interaction severity and management recommendations are not included as they may be highly dependent on the patient and clinical scenario. |
|||
Case: GS is a 40-year-old obese female with type 2 diabetes and generalized anxiety disorder. Her medications include lisinopril 10 mg PO daily, metformin 1000 mg PO BID, sertraline 100 mg PO daily, and diazepam 5 mg PO prn up to TID for management of intermittent episodic acute anxiety. She has reported that her diazepam doesn’t work quickly enough to provide adequate relief for her anxiety. The patient reports that she is trying to adhere to a low-fat diet in an effort to control her weight. Recent labs also show suboptimally controlled diabetes with an A1C of 7.3%. The patient was advised to continue taking metformin with meals and to emphasize healthy foods featuring desirable fats such as nuts, avocado, olive oil, and fatty fish. The patient was advised to take diazepam on an empty stomach whenever possible and to see if it controlled her anxiety symptoms more quickly.
As will be demonstrated with other interactions throughout this article, a thorough understanding of interactions may be used by PCPs as a therapeutic advantage. In the case above, GS was not at goal with her current dose of metformin and not experiencing adequate anxiety relief from the diazepam. She was advised to consume healthy fats with her metformin in order to maximize the effectiveness prior to initiating a second medication (Table 1-2). The dosing strategy of diazepam was modified to be taken on an empty stomach before dose increases or changes in therapy were made. These recommendations were made based on provider understanding of how GI motility affects onset and overall absorption of the respective medications in light of the desired treatment goals.
Another factor that contributes to drug bioavailability is the acidic environment of the stomach. This harsh environment can destroy drugs before they ever make it to the small intestine for absorption, and some drugs must be specially formulated (e.g., delayed-release omeprazole) to reach the small intestine intact. Oral insulin is another example. Insulin is a protein, and is extensively denatured at the low pH of the stomach so that it must be administered parenterally (i.e., IV, IM, SC) to achieve adequate plasma levels. Phase 2 trials are currently underway to evaluate specially formulated oral insulin therapies that overcome this challenge.6 Amphetamines are yet another example. Amphetamines are ionized in the presence of acids like cranberry juice. Coadministration may result in decreased amphetamine exposure, because the amphetamines are more water soluble (hydrophilic) in their ionized form and less able to absorb into tissues. Table 1-3 and 1-4 further describes this type of interaction. A number of online databases are available for providers to use to investigate these and other DDIs as needed in daily practice. For further information on food-drug interactions, consider reading article 1 in the provider resource tool kit (see Table 7).
First Pass
“First pass” refers to the initial passage of a medication, after absorption from the gut, as it passes through the liver to the bloodstream. First pass concludes when a medication or its metabolites enter systemic circulation and is used as a baseline for determining bioavailability. Bioavailability is defined as the fraction of unchanged drug that reaches systemic circulation or the molecular site of action.4 Once absorbed into the cells of the small intestine (i.e., enterocytes), drugs encounter the body’s next line of defense against xenobiotics, the drug metabolism, and transport system. Understanding this system is an important basis for discussing drug interactions. We will be using four illustrations to depict a slightly simplified physiology of drug transport and metabolism in the body. This will facilitate a deeper understanding of the mechanism of drug interactions.
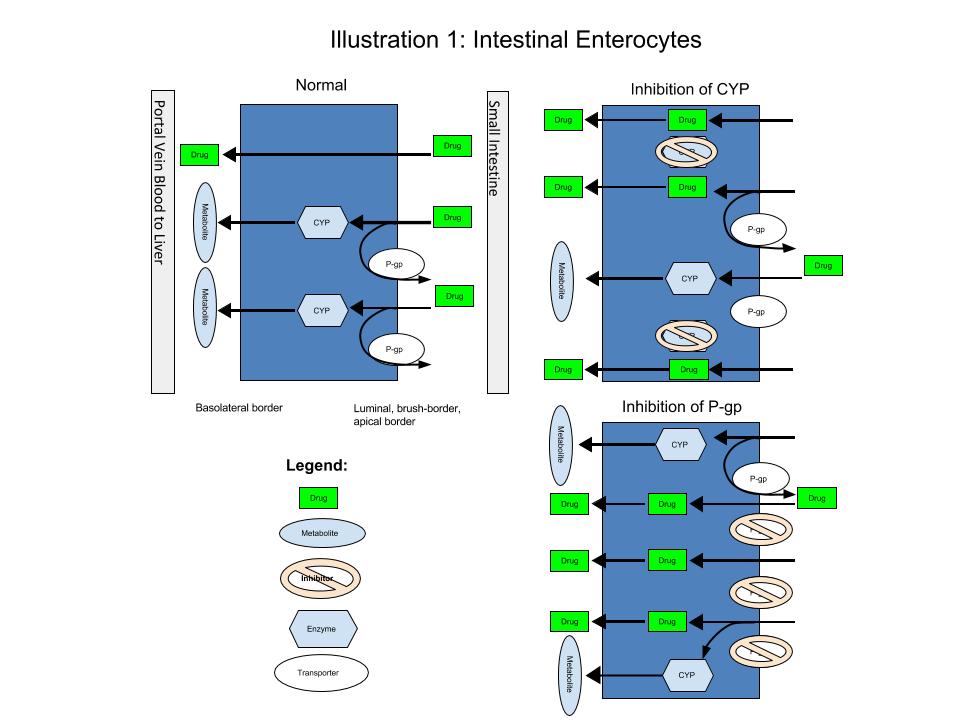
As outlined in Illustration 1, the drug efflux transporter p-glycoprotein (p-gp) and the drug metabolizing enzyme cytochrome P450 3A4 (CYP3A4) are highly expressed in enterocytes. These work together as an intestinal “tag team” (Jessica Oesterheld, MD, e-mail communication, December 2014). Drugs are continually absorbed into the enterocyte. P-gp effluxes the drug molecule out of the enterocyte, where the drug molecules continue to re-enter. Inside the cell, drugs encounter CYP3A4 and undergo extensive metabolism. These processes happen over and over during GI transit so that the amount of drug that reaches the portal vein will be significantly decreased if it is a substrate for either of these pathways. Several examples of interactions that take place at this juncture include HMG-CoA reductase inhibitor (statin) medications and grapefruit, cyclosporine, and diltiazem, and protease inhibitors and ritonavir (see Table 1-6, 1-7 and 1-8).
Understanding Inhibition and Induction
Competitive inhibition. This can function in two ways. One molecule (inhibitor) may have a higher affinity for the metabolic enzyme so that it binds more readily to the enzyme, and the interacting medication (drug) is displaced and metabolized less readily. Another way this type of inhibition occurs is that the inhibitor molecule may have a greater concentration at the site of metabolism, saturating it and outcompeting the interacting medication for the metabolic action of the enzyme. This may be a similar function at the transporter level, but this is less well-understood.
Mechanism-based inhibition. In this type of inhibition, the inhibiting substance (inhibitor) binds irreversibly to the enzyme, rendering the enzyme functionally inert. This removes the potential for the enzyme to metabolize the interacting drug (drug). In theory, the effects of mechanism-based inhibition may last longer. The permanent nature of the alteration of the enzyme requires that more enzyme must be produced by the body to replace the enzyme rendered inert by the inhibiting reaction. The difference between competitive and mechanism-based inhibition may not be clinically significant. The end result is that the medication that is less readily metabolized is more likely to have increased systemic exposure and effect.
Induction. This occurs when a drug signals nuclear receptors of cells to increase expression of a particular drug-metabolizing enzyme. Classic examples of inducers include the enzyme-inducing anticonvulsants phenobarbital, primidone, phenytoin, and carbamazepine. Rifampin is another classic enzyme-inducing drug, and smoking also has significant enzyme-inducing effects. The effects of induction would actually be opposite of the effects seen in Illustration 1. Increased transporter and cytochrome activity would ultimately lead to decreased amounts of drug reaching the bloodstream.
Predicting interactions on the basis of inhibition or induction of transporter proteins can be more challenging than those based on enzymatic inhibition or induction. In practice, providers should rely on pharmacokinetics studies regarding transporter-based interactions. Drug transport is an area of intense investigation with new discoveries and understanding of the clinically relevant interactions still being made.
To adequately understand transporter-based interactions, it is important to have specific knowledge of both the function and location of the transporter proteins involved. For example, inhibition of the effluxing transporter p-gp in the enterocytes (see Illustration 1) will result in increased drug available to reach systemic circulation and, therefore, increased drug exposure. Likewise, inhibition of a hepatic influx transporter, such as SLCO1B1 (see the basolateral border of Illustration 2), will result in less exposure of the drug to CYPs and, therefore, increased systemic exposure due to decreased metabolism. Also, inhibiting influx transporters OAT1 and OAT3 on the basolateral side of the proximal renal tubule will result in increased drug exposure due to decreased renal elimination (see Illustration 3).
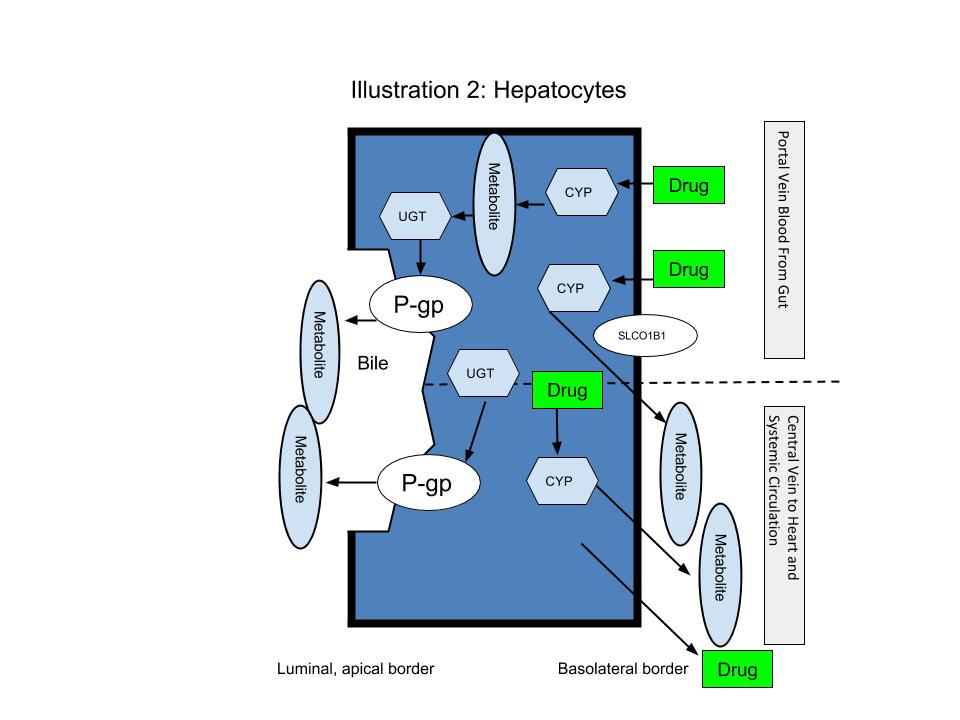
For some drugs that have active metabolites, interactions involving inhibition may shift the metabolic ratio such that less of the active metabolite is formed and effectiveness is lost. When the parent molecule is inert relative to the active metabolite, the drug can be classified as a prodrug. Codeine, clopidogrel, and tamoxifen are three examples of prodrugs. Many drugs fall into a category in which there is activity in both the parent and metabolite form.
As with CYP metabolism, UGT glucuronidation usually inactivates drugs but there are some glucuronide metabolites that are active or more potent than the original parent molecule. The glucuronide metabolite of morphine, morphine-6-glucuronide, is one example of this.12 Table 4 lists some of the of UGT metabolized drugs, inhibitors, and inducers.
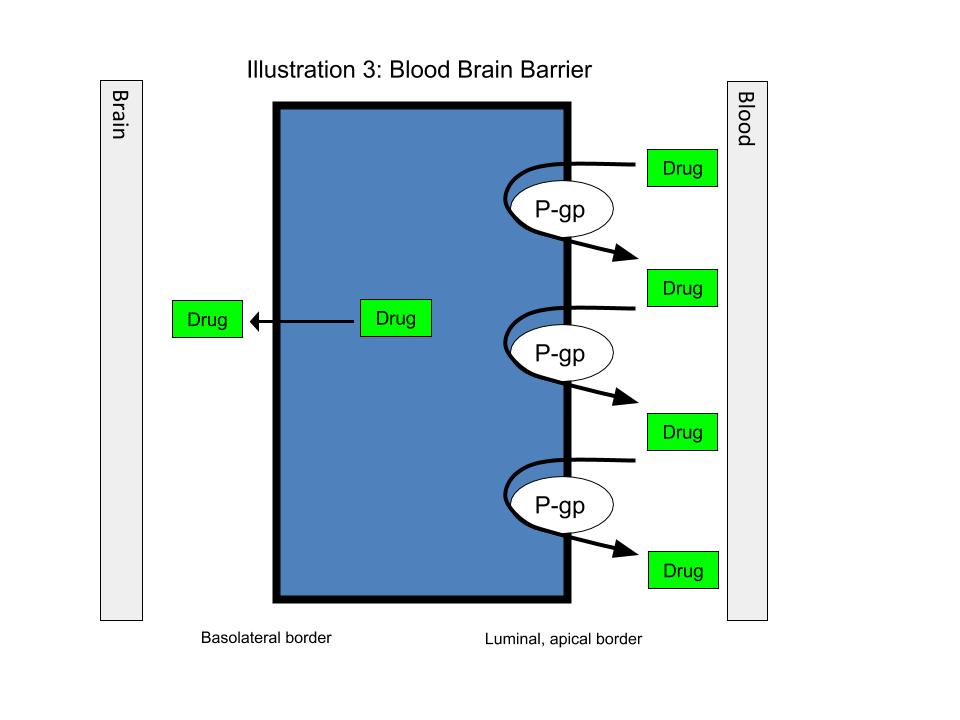
Table 2. Common Drugs by CYP Enzyme
|
CYP2D6 |
CYP2C9 |
CYP2C19 |
CYP3A4/5 |
|
Antiarrhythmics Antidepressants Antipsychotics Beta-blockers Opioids |
NSAIDs Phenytoin Sulfonylureas Valproic acid Warfarin |
Antidepressants Carisoprodol Diazepam Plavix
Proton pump |
Benzodiazepines
Hormone Corticosteroids Immunosuppressants Protease inhibitors Statins Transplant medications |
Some transporters, which are not discussed in this article, can result in decreased exposure to a drug when they are inhibited. SLCO2B1, an influx transporter expressed in enterocytes (luminal border), represents an example of this type of interaction. Substances in orange juice act as inhibitors of SLCO2B1. Aliskiren, an antihypertensive renin inhibitor, is a substrate of this transporter. Coadministration of aliskiren and orange juice resulted in up to a 62% decrease in aliskiren exposure and an 80% decrease in Cmax.7
In theory, inhibition of p-gp efflux in hepatocytes could actually result in a “trapping” of the drug in the hepatocyte and increased metabolism of the drug, resulting in decreased systemic exposure, rather than the drug entering into the bile and undergoing enterohepatic recirculation. Lastly, inhibition of the efflux transporter p-gp in the blood-brain barrier (BBB) would not be expected to change systemic exposure of a drug to any clinically significant extent, but it may increase the amount of drug that reaches the CNS and, therefore, increase the risk of toxicity. The difference between these potential effects and the effect of p-gp inhibition in the enterocyte illustrates the importance of considering the location of action as well as the affected transporter in anticipating the clinical impact of inhibition or induction on transporter-based interactions.
Phase I Drug Metabolism
Once beyond the gut, drugs and their respective metabolites enter the portal vein en route to the liver. Here they are metabolized through the hepatocytes before they either enter back into systemic circulation via the central vein or are cleared via the bile. Drug molecules can enter hepatocytes by transporters, as already discussed, or by other means, such as passive diffusion (see Illustration 2). For drugs that rely on transporters, like statins, clinically significant interactions may exist if their influx transporter (SLCO1B1) is inhibited. In this case, the blood levels of the statin may significantly increase. This would increase the risk for adverse effects, such as myalgias and rhabdomyolysis (see Table 3-8).
Highly expressed as well as highly polymorphic, the various families of cytochrome P450 enzymes (CYPs) are also encountered within the hepatocytes. Although the discussion will focus on these enzymes, it is important to note that there are many others that metabolize drugs during phase I and phase II metabolism (see Figure 3). However, CYPs are among the most studied and well understood in terms of clinically significant interactions.
Figure 3. Phase I and Phase II Drug Metabolism
Adapted from: Yang Z, Yu Y, Yao L, et al. DetoxiProt: an integrated database for detoxification proteins. BMC Genomics 2011;12(Suppl 3):S2.
Phase I metabolism is the term used to describe the process of adding or exposing hydrophilic functional groups (functionalization) to drug molecules.1 From a drug interaction standpoint, CYP2D6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5 are the most important, as they are involved in the metabolism of the majority of all hepatically cleared drugs.8 Examples of common drugs and drug classes metabolized by these pathways can be found in Table 2. The types of reactions that cytochromes have with drugs is oxidative in nature. Reactions can result in inactivation or activation of drugs as well as a change in structure that makes the drug molecule more amenable to conjugation via phase II drug-metabolizing enzymes and/or more amenable to elimination. Because so many medications are metabolized by the CYP450s and because these enzymes can dramatically influence the exposure of drugs, this system is highly vulnerable to interactions. One useful example to illustrate the impact of this mechanism is the interaction between dextromethorphan and quinidine.
Table 3. Hepatic Interaction Examples
|
Drug Combination |
Mechanism |
Effect |
|
|
1 |
dextromethorphan + quinidine |
CYP2D6 & P-gp inhibition |
Dextromethorphan/ quinidine 30 mg/10 mg PO BID resulted in up to a 20-fold increase in dextromethorphan exposure compared to dextromethorphan alone.1 |
|
2 |
warfarin + amiodarone |
CYP2C9 inhibition |
Warfarin and amiodarone resulted in a 55% decrease in warfarin clearance.2 |
|
3 |
citalopram + omeprazole |
CYP2C19 inhibition |
Multidosed omeprazole 20 mg daily x 18 days and single dosed citalopram 20 mg on day 8 resulted in a 120% increase to s-citalopram plasma concentrations and no statistically significant change for r-citalopram.3 |
|
4 |
ethinyl estradiol/ levonorgestrel + phenytoin |
CYP3A4 induction |
Multidosed phenytoin 200 mg to 300 mg daily x 8 to 12 weeks and single-dosed ethinyl estradiol 50 mcg/levonorgestrel 250 mcg resulted in a 49% decrease in ethinyl estradiol AUC and a 42% decrease in levonorgestrel AUC.4 |
|
5 |
clozapine + cigarette smoking |
CYP1A2 induction |
Smoking and clozapine resulted in 50% decrease in clozapine levels, and when smoking was discontinued up to 300% increase in clozapine levels have been reported.5,6 |
|
6 |
alcohol + disulfiram |
aldehyde dehydrogenase (ALDH) inhibition |
Disulfiram and its active metabolite inhibit the conversion of acetaldehyde to acetate through inhibition of ALDH. Acetaldehyde blood levels increase five to ten-fold. Symptoms of hyperacetaldehydemia include flush, vasodilatation, throbbing head and neck pain, respiratory difficulties, vomiting, sweating, and thirst.7 |
|
7 |
methanol or ethylene glycol + ethanol or fomepizole |
alcohol dehydrogenase (ADH) inhibition |
Ethanol and fomepizole both serve as an antidote by competitively inhibiting the metabolism of methanol or ethylene glycol at ADH resulting in the decreased formation of either of these drugs to their toxic metabolites.8,9 |
|
8 |
methylphenidate + carbamazepine |
CES1 induction |
Coadministration has resulted in several cases of decreased methylphenidate exposure and decreased effectiveness.10,11 |
|
9 |
rosuvastatin + gemfibrozil |
SLCO1B1 inhibition |
Multidosed gemfibrozil 600 mg BiD x 7 days and single-dosed rosuvastatin 80 mg resulted in an 88% increase in rosuvastatin AUC and a 121% increase in Cmax.12 |
|
10 |
rosuvastatin + cyclosporine |
SLCO1B1 inhibition |
Multidosed cyclosporine 75 mg to 200 mg BiD and multidosed rosuvastatin 10 mg to 20 mg daily x 10 days resulted in a 610% increase to rosuvastatin AUC and a 960% increase to rosuvastatin Cmax.13 |
|
11 |
cyclosporine + repaglinide |
SLCO1B1 + CYP3A4 inhibition |
Multidosed cyclosporine 100 mg daily x 2 days and single-dosed repaglinide 0.25 mg resulted in an 140% increase in repaglinide AUC and 80% increase in Cmax. This may enhance the blood glucose-lowering effects of repaglinide and increase the risk of hypoglycemia.14 |
|
12 |
digoxin + ritonavir |
P-gp inhibition |
Multidosed ritonavir 300 mg BiD x 11 days and single-dosed digoxin IV 500 mcg resulted in an 86% increase to digoxin AUC.15 |
|
13 |
lamotrigine + ethinyl estradiol |
UGT2B7 induction |
Multidosed ethinyl estradiol 30 mcg/ levonorgestrel 150 mcg daily and multidosed lamotrigine 300 mg daily resulted in a 52% decrease in lamotrigine AUC and 39% decrease in lamotrigine Cmax.16 There have been cases of breakthrough seizures with this combination.17 |
|
14 |
zidovudine + probenecid |
UGT2B7 inhibition |
Coadministration resulted in a 106% increase in zidovudine AUC.18 |
|
15 |
lorazepam + valproic acid |
UGT2B7 & UGT2B15 inhibition |
Coadministration has resulted in a 20% to 67% increase in lorazepam AUC.19,20 |
|
16 |
valproic acid + ethinyl estradiol |
UGT2B7 induction |
Coadministration has resulted in valproic acid plasma concentration decreases of up to 46% with increased seizure frequency in one report.21,22,23 |
|
17* |
azathioprine + sulfasalazine |
TPMT inhibition |
Multidosed sulfasalazine mean dose 2.1 grams daily and multidosed azathioprine mean dose 92.8 mg daily resulted in a 55% discontinuation rate due to rash, gastrointestinal upset, leucopenia, and nephrotic syndrome.24 |
|
Tables may not include entire mechanisms or studies with conflicting findings. Interaction severity and management recommendations are not included as they may be highly dependent on the patient and clinical scenario. *TPMT is a phase II enzyme but its location may not be primarily hepatic. |
|||
|
1. Nuedexta® [package insert]. Avanir Pharmaceuticals, Inc. Aliso Viejo, CA; December 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=484e0918-3442-49dc-8ccf-177f1f3ee9f3 Accessed December 29, 2014.. 2. Almog S, Shafran N, Halkin H, et al. Mechanism of warfarin potentiation by amiodarone: dose—and concentration—dependent inhibition of warfarin elimination. Eur J Clin Pharmacol 1985;28:257-261. 3. Rocha A, Coelho EB, Sampaio SA, Lanchote VL. Omeprazole preferentially inhibits the metabolism of (+)-(S)-citalopram in healthy volunteers. Br J Clin Pharmacol 2010;70:43-51. 4. Crawford P, Chadwick DJ, Martin C, et al. The interaction of phenytoin and carbamazepine with combined oral contraceptive steroids. Br J Clin Pharmacol 1990;30:892-896. 5. Derenne JL, Baldessarini RJ. Clozapine toxicity associated with smoking cessation: case report. Amer J Ther 2005;12:469-471. 6. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol 2006;62:1049-1053. 7. Disulfiram [package insert]. Teva Pharmaceuticals, Sellersville, PA; August 2014. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=f0ca0e1f-9641-48d5-9367-e5d1069e8680. Accessed December29, 2014. 8. Fomepizole [package insert]. Paladin Labs Inc., Dover, Del; July 2007. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c045b5a8-b220-4f0a-a831-155b29c19454. Accessed December 29, 2014. 9. Rietjens SJ, de Lange DW, Meulenbelt J. Ethylene glycol or methanol intoxication: which antidote should be used, fomepizole or ethanol? Netherlands J Med 2014;72:73-79. 10. Behar D, Schaller J, Spreat S. Extreme reduction of methylphenidate levels by carbamazepine. J Am Acad Child Adolesc Psych 1998;37:1128-1129. 11. Schaller JL, Behar D. Carbamazepine and methylphenidate in ADHD. J Am Acad Child Adolesc Psych 1999;38:112-113. 12. Schneck DW, Birmingham BK, Zalikowski JA, et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther 2004;75:455-463. 13. Simonson SG, Raza A, Martin PD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther 2004;76:167-177. 14. Cyclosporine [package insert]. Sandoz Inc., Princeton, NJ; September 2014. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=8893395a-6bfa-4b3c-84c2-d1d0c923304f Accessed December 30, 2014. . 15. Ding R, Tayrouz Y, Riedel KD, et al. Substantial pharmacokinetic interaction between digoxin and ritonavir in healthy volunteers. Clin Pharmacol Ther 2004;76:73-84. 16. Sidhu J, Job S, Singh S, Philipson R. The pharmacokinetic and pharmacodynamic consequences of the co-administration of lamotrigine and a combined oral contraceptive in healthy female subjects. Br J Clin Pharmacol 2006;61:191-199. 17. Sabers A, Buchholt JM, Uldall P, Hansen EL. Lamotrigine plasma levels reduced by oral contraceptives. Epilepsy Res 2001;47:151-154. 18. Zidovudine [package insert]. GlaxoSmithKline, RTP, NC; November 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=360d8a6d-d3b6-43ef-8764-e99d831880a4 Accessed December 29, 2014. 19. Samara EE, Granneman RG, Witt GF, Cavanaugh JH. Effect of valproate on the pharmacokinetics and pharmacodynamics of lorazepam. J Clin Pharmacol 1997;37:442-450. 20. Anderson GD, Gidal BE, Kantor ED, Wilensky AJ. Lorazepam-valproate interaction: studies in normal subjects and isolated perfused rat liver. Epilepsia 1994;35:221-225. 21. Herzog AG, Blum AS, Farina EL, et al. Valproate and lamotrigine level variation with menstrual cycle phase and oral contraceptive use. Neurology 2009;72:911-914. 22. Herzog AG, Farina EL, Blum AS. Serum valproate levels with oral contraceptive use. Epilepsia 2005;46:970-971. 23. Galimberti CA, Mazzucchelli I, Arbasino C, et al. Increased apparent oral clearance of valproic acid during intake of combined contraceptive steroids in women with epilepsy. Epilepsia 2006;47:1569-1572. 24. Helliwell PS. Combination therapy with sulphasalazine and azathioprine. Br J Rheumatol 1996;35:493-494. |
|||
Dextromethorphan, an opioid analog that is commonly used to treat cough,4 was recently discovered to be an effective medication for pseudobulbar affect, a neurologic disorder that can increase emotional instability.9 Dextromethorphan is also a CYP2D6 and p-gp substrate. To be effective at treating pseudobulbar affect, high concentrations of the drug must pass through the BBB and into the CNS. For most individuals, this is not possible without giving very high doses of dextromethorphan. Coadministration with low-dose quinidine, a potent CYP2D6 inhibitor, boosts systemic exposure of dextromethorphan up to 20-fold (see Table 3-1). Illustration 2 can help the reader visualize how inhibition of the p-gp and CYP2D6 enzymes could result in significantly increased exposure of dextromethorphan in the blood. An interesting fact about this interaction is that coadministration with quinidine is not required in individuals who are genetically deficient in the CYP2D6 enzymes (CYP2D6 poor metabolizers). This is because dextromethorphan exposure is sufficiently high in these patients. Exposing them to quinidine, which has its own profile of adverse effects such as QTc prolongation, is unnecessary.10
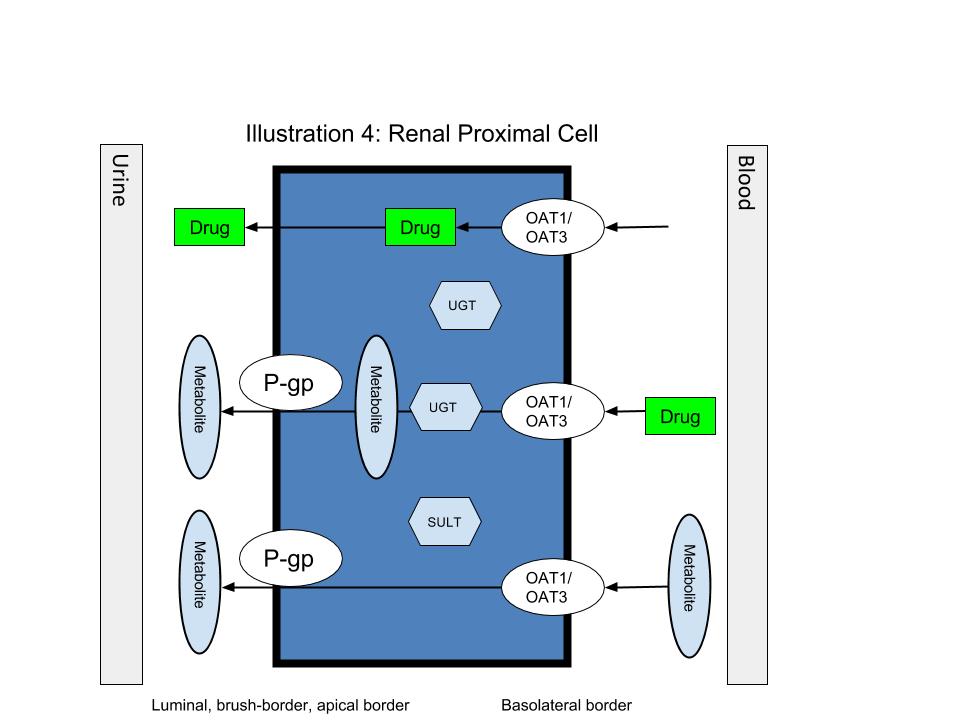
Illustration 4 can be used to visualize how this particular combination may interact at the BBB. With quinidine inhibiting the p-gp protein at this location, dextromethorphan is less rapidly effluxed from the brain. Thus, dextromethorphan is able to reach therapeutic effectiveness without requiring large doses.
Table 4. Examples of UGT Substrates, Inhibitors, and Inducers34,35
|
UGT1A1 |
UGT2B7 |
UGT2B15 |
|
Substrates Carvedilol Estradiol Ezetimibe
Irinotecan’s active Inhibitors Atazanavir Ketoconazole Lopinavir/ritonavir Inducers Carbamazepine Curcuminoids Fenofibric acid Methimazole Phenobarbital Phenytoin Progesterone Rifampin Ritonavir St. Johns wort |
Substrates Buprenorphine Carvedilol Codeine Cyclosporine Diclofenac Efavirenz Lamotrigine Lorazepam Morphine Mycophenolate acid Naloxone Naproxen Inhibitors Fluconazole Ketoconazole Medroxyprogesterone Probenecid Valproic acid Inducers Ethinyl estradiol Lopinavir/ritonavir Nelfinavir Phenobarbital Rifampin |
Substrates Lorazepam Oxazepam Inhibitors Ibuprofen Ketoconazole Soybean extract Tacrolimus Inducers Carbamazepine Phenobarbital Phenytoin Rifampin |
Case: BD is an 85-year-old male nursing home patient who has been emotionally labile following a stroke he experienced about 1 month ago, according to staff reports. The physician diagnosed the patient with pseudobulbar affect and inquired with the consulting pharmacist on appropriate drug regimens for the patient. Pharmacogenetic testing had previously been ordered on this patient due to multiple comorbidities and recognized polypharmacy. He is also known to be a CYP2D6 poor metabolizer. The pharmacist suggested that while a newer combination drug of dextromethorphan/quinidine (Nuedexta) is indicated for pseudobulbar affect, the medication is not recommended in CYP2D6 poor metabolizers since the exposure to quinidine is unnecessary to produce a metabolic suppression that is already present in those patients. The pharmacist recommended initiating dextromethorphan at 20 mg PO daily for 1 week and then increasing to 20 mg PO BID on day 8 thereafter.
As shown in this case, knowledge of the mechanism of drug interactions can be used as a therapeutic advantage to successfully treat disease. Understanding why the quinidine component was unnecessary in this patient allowed for a safer therapeutic plan in a patient with recognized polypharmacy. Other examples of Phase I interactions are included in Table 3 and include the use of disulfiram to deter alcoholics from drinking (see Table 3-6) and the use of ethanol or fomepizole to treat methanol and ethylene glycol poisoning (see Table 3-7).
Case: MB is a 62-year-old female and heavy drinker. She calls the PCP’s office asking for advice on treatment of bacterial vaginosis. Based on her known history of alcoholism, the physician discourages the use of OTC intravaginal metronidazole and offers to call in a prescription for clindamycin phosphate vaginal cream.
Metronidazole has resulted in cases of disulfiram-like reactions with alcohol. This is likely due to metronidazole-possessing aldehyde dehydrogenase (ALDH)-inhibiting properties. This increases plasma levels of acetaldehyde and causes unpleasant side effects: flush, vasodilatation, throbbing head and neck pain, respiratory difficulties, vomiting, sweating, and thirst. Although this interaction would probably be less likely with intravaginal administration of metronidazole, there has been a case report of such an interaction. In the above case, the knowledge of the mechanism of the interaction and assessment of the patient’s long history of alcoholism allowed the provider to choose a treatment regimen less likely to result in adverse effects.11
Phase II Drug Metabolism
Phase II metabolism is the term used to describe secondary drug metabolism and is usually conjugative in nature.1 This usually occurs after phase I metabolism, but this is not always the case. It may happen in the same cell or tissue immediately after phase I metabolism. It may also occur after the drug has re-entered systemic circulation and again entered into tissues like the liver for further metabolism.
The most important phase II enzymes from a drug metabolism and interaction standpoint are the uridine 5’- diphosphate glucuronosyltransferases (UGTs). This enzyme family functions via glucuronidation — the addition (or conjugation) of glucuronic acid, which is highly water soluble — to a drug molecule.
Illustration 2 can be used to visualize the mechanisms of interactions that occur in phase II metabolism. Lamotrigine is a non-enzyme inducing anticonvulsant that undergoes glucuronidation to inactive metabolites. The known UGTs that metabolize lamotrigine include UGT2B7, UGT1A3 and UGT1A4. Ethinyl estradiol is known to induce glucuronidation and has been shown to result in clinically significant decreases in lamotrigine blood levels (see Table 3-13). Using Illustration 2, one can infer that the induction of UGT metabolism of lamotrigine with ethinyl estradiol could result in increased conversion of lamotrigine to inactive metabolites. This would decrease blood levels of the active drug and, therefore, decrease effectiveness.
Case: CL is a 32-year-old female who requests a prescription for oral contraception. She recently got married and does not plan on having children. She has a seizure disorder and is being treated with lamotrigine 150 mg PO BID. The provider began issuing a prescription for birth control via the electronic health record when an interaction alert flagged stating that blood levels of lamotrigine may be decreased due to UGT induction by ethinyl estradiol. The provider researched and found no such interaction with progestin-only contraceptives and a prescription for norethindrone 0.35 mg PO daily was considered. After counseling and discussion, due to higher potential for pregnancy with progestin-only oral contraception in the presence of a potential teratogen in lamotrigine, the patient elected to implement a levonorgestrel-releasing intrauterine system as a primary method of birth control.
Thiopurine methyltransferase (TPMT) is another example of a phase II drug-metabolizing enzyme, although it is unique in that it conjugates drugs with methyl groups (methylation), which is a process that actually decreases water solubility. It is important for the inactivation of thiopurine drugs like azathioprine, 6-mercaptopurine, and 6-thioguanine.
Case: JW is a 38-year-old female with Crohn’s disease who presented for recent labs. Her prescribed medications include azathioprine 2 mg/kg/day and a combined oral contraceptive. Her labs returned as follows: WBC 3000 cells/mm3 (3200-9800); Hgb 10% (12-16); Hct 33% (33-43); PLT 130,000/ mm3 (130,000-400,000). Further discussion with the patient revealed that she was consuming large amounts of OTC naproxen sodium for increasing joint pain. The patient was advised to immediately discontinue naproxen, and the dose of azathioprine was reduced to 1 mg/kg/day until the CBC indices normalized. The patient was issued a prescription for tramadol as needed for joint pain to be taken instead of naproxen.
The two cases above illustrate the importance of understanding phase II metabolism interactions as thoroughly as phase I interactions. Although the known interactions are fewer in numbers than phase I interactions, their clinical importance should not be overlooked. Article 3 in the provider resource tool kit (see Table 7) provides a comprehensive review of the enzymes covered here, as well as others that are involved in drug metabolism. Article 4 provides a specific review on UGTs.
Distribution
Although this article will not go into a great deal of review on drug distribution, this is also an important concept for understanding drug interactions. As a drug reaches systemic circulation, it will begin to distribute into tissues. As with absorption, distribution of a drug is dependent on a host of factors, such as its blood concentration (a direct result of factors such as dose, bioavailability, and clearance), protein binding, lipophilicity (fat solubility), the patient’s body weight, fat distribution, and renal function.
Table 5. Distribution Interaction Examples
|
Drug Combination |
Mechanism |
Effect |
|
|
1. Sadeque AJ, Wandel C, He H, et al. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther 2000;68:231-237. 2. Valproic acid [package insert]. Catalent Pharma Solutions, LLC., St. Petersburg, FL; October 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=cef3e335-5891-412f-89c4-79fcd2f145b7 Accessed December 30, 2014. 3. Tollefson GD. Delirium induced by the competitive interaction between phenytoin and dipropylacetate. J Clin Psychopharmacol 1981;1:154-158. 4. Yoon HW, Giraldo EA, Wijdicks EF. Valproic acid and warfarin: an underrecognized drug interaction. Neurocrit Care 2011;15:182-185. |
|||
|
1 |
loperamide + quinidine |
p-gp inhibition |
Loperamide 16 mg and quinidine 600 mg resulted in statistically significant respiratory depression compared to loperamide alone.1 |
|
2 |
phenytoin + valproic acid |
albumin binding displacement |
A complex interaction. Valproate 400 mg TID and phenytoin 250 mg resulted in a 60% increase in the free fraction of phenytoin,2 which may be associated with transient increases in phenytoin toxicity.3 |
|
3 |
warfarin + valproic acid |
albumin binding displacement |
Valproic acid loading dose may displace warfarin from protein binding site increasing the free unbound warfarin levels and increase INR to a clinically significant extent.3 |
|
Tables may not include entire mechanisms or studies with conflicting findings. Interaction severity and management recommendations are not included as they may be highly dependent on the patient and clinical scenario. |
|||
Interactions involving distribution tend to be more difficult to identify because they don’t necessarily result in a change to the systemic exposure of the drug. One type of distribution-based interaction affects how much of the free drug (i.e., unbound from plasma protein) is available to distribute into tissues and/or bind to a molecular site of action (bound drug is considered unavailable for either of these). The interaction between phenytoin and valproic acid is a classic example that demonstrates this (see Table 5-2).
Interactions that deal with protein displacement are actually controversial, and some experts claim that any change in unbound free drug by protein displacement would be buffered by redistribution, metabolism, and elimination of the newly freed/unbound drug.13 According to one expert, for a protein displacement interaction to be clinically significant, it must meet the following criteria: the victim drug must be > 90% protein-bound, have a narrow therapeutic index, have a high hepatic extraction ratio, and be administered as an IV formulation.13
Another distribution type of interaction is that between loperamide and quinidine (see Table 5-1). Loperamide is an antidiarrheal agent with potent mu opioid binding affinity but poor penetration into the central nervous system.4 Under normal circumstances, loperamide is effluxed out of the brain by p-gp and does not have a significant CNS effect. However, when combined with a p-gp inhibitor like quinidine, the loperamide may begin to accumulate in the brain, resulting in adverse effect and toxicity.
Case: MB is a 65-year-old female with the stomach flu. She is being treated with quinidine 200 mg PO QID for maintenance of sinus rhythm. She sent her son to the pharmacy to get over the counter loperamide for treatment of her diarrhea and immediately started taking the maximum dose of 4 mg PO initially and then 2 mg PO after each loose stool. The next day, she asked her son to drive her to the minor care clinic where she checked in with complaints of nausea, vomiting, headache, dizziness, and fatigue.
This case also demonstrates the importance of clearly instructing patients to check with their PCPs and pharmacists prior to taking new medications, even those available over the counter.
Excretion
As drugs and their respective metabolites circulate, they are eventually eliminated from the body. The primary elimination site for most drugs is the kidneys, but other elimination routes include the hepatobiliary system, the lungs, skin, saliva, and breast milk. Providers commonly adjust for delays in renal elimination by estimating the glomerular filtration rate via use of the creatinine clearance calculation. This estimate is used to dose many drugs, especially antibiotics. However, anticipating dose adjustments for drug interactions that take place at the kidneys may be less familiar to most providers. Illustration 3 depicts the simplified physiology of renal tubular secretion in the kidneys. Even more so than the other sites covered in this article, renal clearance is a complex process and this review only addresses the highlights of this system. For more information on this system, consider reading article 7 in the provider resource tool kit (see Table 7).
Table 6. Kidney Elimination Interaction Examples
|
Drug Combination |
Mechanism |
Effect |
|
|
1. Probenecid [package insert]. Lannett Company, Inc. Philadelphia, PA; November 2010. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=ab497fd8-00c3-4364-b003-b39d21fbdf38 Accessed December 28, 2014. 2. Hill G, Cihlar T, Oo C, et al. The anti-influenza drug oseltamivir exhibits low potential to induce pharmacokinetic drug interactions via renal secretion-correlation of in vivo and in vitro studies. Drug Metab Dispos 2002;30:13-19. 3. Cidofovir [package insert]. Mylan Institutional, Rockford, IL; November 2012. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=56541229-8c1a-4550-8951-2415ed08e7e9 Accessed December 29, 2014. 4. Inotsume N, Nishimura M, Nakano M, et al. The inhibitory effect of probenecid on renal excretion of famotidine in young, healthy volunteers. J Clin Pharmacol 1990;30:50-56. |
|||
|
1 |
penicillin + probenecid |
OAT3 inhibition |
Coadministration results in a 100% to 300% increase in penicillin plasma levels.1 |
|
2 |
oseltamivir + probenecid |
OAT1 inhibition |
Single-dosed oseltamivir 150 mg and multidosed probenecid 500 mg resulted in a 150% increase in oseltamivir active metabolite AUC and 100% increase in Cmax.2 |
|
3 |
cidofovir + probenecid |
OAT1 inhibition |
Probenecid and cidofovir 5 mg/kg resulted in a 44% increase in cidofovir AUC and a 34% decrease in cidofovir renal clearance.3 |
|
4 |
famotidine + probenecid |
OAT3 inhibition |
Multidosed probenecid 1500 mg and single-dosed famotidine 20 mg resulted in an 81% increase in famotidine AUC.4 |
|
Tables may not include entire mechanisms or studies with conflicting findings. Interaction severity and management recommendations are not included as they may be highly dependent on the patient and clinical scenario. |
|||
During kidney elimination, many drugs will be passively filtered through the glomerulus and eliminated this way. However, many drug/metabolite molecules also undergo renal tubular secretion, which is an extremely important site for drug interactions since this is a predominantly transporter-mediated process.14 Renal tubular secretion happens in the proximal tubule of nephrons and again involves a highly complex network of transporters, some of them energy dependent, that move drugs and other electrolytes in and out of the collecting tubule (urine) and blood. Glucuronidation and sulfation (via sulfotransferases [SULTs]) also occur in the renal proximal tubule. This is an area of drug metabolism that requires further investigation, but it is believed that the contribution to overall drug metabolism in the kidneys is much less clinically significant compared to that which occurs in the intestine and liver.14
Two of the most well-understood transporters from a drug interaction standpoint are the organic anion transporters, OAT1, and OAT3. These transporters work by exchanging intracellular endogenous molecules with extracellular anions.15 Probenecid, used as an antigout agent due to its ability to inhibit reabsorption of uric acid from the collecting tubule, is a classic inhibitor of these transporters.
Table 7. Provider Resource Tool Kit
|
1 |
Review of food and drug interactions |
Winstanley PA, Orme ML. The effects of food on drug bioavailability. Br J Clin Pharmacol 1989;28:621-628. |
|
2 |
Review of gut wall interactions |
Thelen K, Dressman JB. Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol 2009;61:541-558. |
|
3 |
Comprehensive review of metabolic pathways and drug interactions |
Wynn G, Oesterheld J, Cozza K, Armstrong S. Clinical Manual of Drug Interaction Principles for Medical Practice. Arlington, VA: American Psychiatric Publishing, Inc.; 2009. |
|
4 |
Review of UGTs |
Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 2013;45:1121-1132. |
|
5 |
Discussion of protein binding displacement interactions |
Rolan PE. Plasma protein binding displacement interactions—why are they still regarded as clinically important? Br J Clin Pharmacol 1994;37:125-128. |
|
6 |
Review of transporters |
Han HK. Role of transporters in drug interactions. Arch Pharm Res 2011;34:1865-1877. |
|
7 |
Review of kidney based drug elimination |
Tett SE, Kirkpatrick CM, Gross AS, McLachlan AJ. Principles and clinical application of assessing alterations in renal elimination pathways. Clin Pharmacokinet 2003;42:1193-1211. |
Case: An elderly gentleman with gout and COPD presents with flu-like symptoms and tests positive for influenza. His wife is also a former smoker with COPD. Oseltamivir 75 mg PO BID x 5 days is prescribed for the husband for treatment and oseltamivir 75 mg PO daily x 10 days is prescribed for the wife for prophylaxis. Due to a recent flu outbreak they discovered that the pharmacy only has 15 tablets of oseltamivir in stock. Knowing that the husband is taking probenecid to control gout, the prescription for the husband is changed to 75 mg one tablet daily x 5 days and the wife is continued on 75 mg one tablet daily x 10 days.
This case demonstrates how inhibition of the OAT transporters may result in clinically significant increases in blood levels of drugs (see Figure 4). As with the drug-metabolizing enzymes, by understanding the drug’s route of elimination, providers can avoid harmful interactions and sometimes promote beneficial ones.
Figure 4.
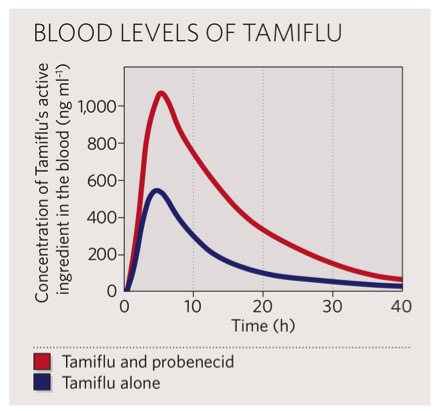
Adapted from: Butler D. Wartime tactic doubles power of scarce bird-flu drug. Nature 2005;438:6.
Summary and Discussion
As pharmaceutical sciences advance, one certainty is that the constellation of medications that PCPs will be expected to track and manage in their role as coordinators of patient care will continue to expand. To further complicate this issue, aging patient populations will only increase the incidence of comorbidities that make polypharmacy a frequent concern for the PCP. While software tools can help facilitate the recognition of current or potential DDI in the polypharmacy patient, the software cannot always provide adequate information about managing DDI while still meeting treatment goals.
The best solution to this DDI dilemma is to seek out and capitalize on the expanding knowledge of the fundamental mechanisms of drug metabolism. This understanding can accomplish much more than simply avoiding DDI or mitigating its dangers and effects when it is encountered. Some of the cases presented here have shown that an understanding of metabolic processes can be leveraged to create opportunities for achieving treatment goals in the face of multiple challenges. This can help achieve treatment goals not only in the face of DDI, but complicating situations like temporary pharmaceutical shortages or the presence of genetic variations in the patient that affect drug metabolism.
It is important to remember that community resources, especially pharmacists, have more extensive training that can and should be fully utilized by PCPs to facilitate understanding and management of pharmaceutical therapies, including metabolism-related DDI. Developing protocols that allow physicians and their patients to make better and more reliable use of these resources represents a powerful tool for the PCP.
A potential example could be found in dealing with one of the frustrations PCPs face in managing pharmaceutical treatment: the formulary requirements of insurance payers. This often presents obstacles when trying to prescribe safer medications based on DDI or combine drugs in unique ways to optimize pharmacotherapy. This presents an opportunity for primary care providers and pharmacists to develop cooperative protocols and initiatives. The emergence of the PCMH model of care in recent years presents an ideal framework for this cooperative approach to patient care. By seeking tools that allow PCPs to integrate pharmacist input into patient care decision-making, PCPs will be better able to leverage the additional training and expertise that pharmacists can bring to complex issues of medication management. Working together, PCPs and pharmacists could incorporate the formulary recommendations from insurance payers using sound clinical reasoning rather than simply responding to financial pressures. To facilitate this, software could be developed that identifies alternative medications not only on the basis of formulary approval but also incorporating the identification of potential risks.
While developing this understanding is certainly useful for the PCP, it should be emphasized that this subject is incredibly complex and developing rapidly. New information emerges constantly as part and parcel of the research engines that drive the pharmaceutical industry. The example cases and accompanying illustrations in this review have sometimes been simplified in order to demonstrate the basic principles of drug metabolism and transport, but should not be regarded as a comprehensive or in-depth review.
References
1. Gonzalez FJ, Nebert DW. Evolution of the P450 gene superfamily: Animal-plant ‘warfare,’ molecular drive and human genetic differences in drug oxidation. Trends Gen 1990;6:182-186.
2. Glassman PA, Simon B, Belperio P, Lanto A. Improving recognition of drug interactions: Benefits and barriers to using automated drug alerts. Med Care 2002;40:1161-1171.
3. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: A postal survey of US prescribers. Drug Safety 2008;31:525-536.
4. Brunton LL, Chabner BA, Knollmann BC, eds. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York: McGraw-Hill; 2011.
5. Winstanley PA, Orme ML. The effects of food on drug bioavailability. Br J Clin Pharmacol 1989;28:621-628.
6. Oramed Pharmaceuticals, Inc. June 2014. Available at http://oramed.com/userfiles/files/0801-clinical-trial-chart - 15_06_14.pdf. Accessed January 2, 2015.
7. Tapaninen T, Neuvonen PJ, Niemi M. Orange and apple juice greatly reduce the plasma concentrations of the OATP2B1 substrate aliskiren. Br J Clin Pharmacol 2011;71:718-726.
8. Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol Sci 2004;25:193-200.
9. Rosen H. Dextromethorphan/quinidine sulfate for pseudobulbar affect. Drugs Today 2008;44:661-668.
10. Nuedexta® [package insert]. Avanir Pharmaceuticals, Inc. Aliso Viejo, CA; December 2013. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=484e0918-3442-49dc-8ccf-177f1f3ee9f3. Accessed December 29, 2014. .
11. Plosker GL. Possible interaction between ethanol and vaginally administered metronidazole. Clin Pharm 1987;6:189-193.
12. Christrup LL. Morphine metabolites. Acta Anaesthes Scandinavica 1997;41(1 Pt 2):116-122.
13. Rolan PE. Plasma protein binding displacement interactions — why are they still regarded as clinically important? Br J Clin Pharmacol 1994;37:125-128.
14. Tett SE, Kirkpatrick CM, Gross AS, McLachlan AJ. Principles and clinical application of assessing alterations in renal elimination pathways. Clin Pharmacokinet 2003;42:1193-1211.
15. Burckhardt G, Wolff NA, Bahn A. Molecular characterization of the renal organic anion transporter 1. Cell Biochem Biophys 2002;36:169-174.
MONOGRAPH: Drug-to-drug interactions are a concern of increasing significance at all levels of health care.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.