Stroke Mimics: A Clinical Dilemma
Authors
John W. Martel, MD, PhD, Assistant Professor, Tufts University School of Medicine, Maine Medical Center, Portland, ME.
Micah Ownbey, MD, Maine Medical Center, Portland, ME.
Calvin Simmons, MD, Maine Medical Center, Portland, ME.
Peer Reviewer
Todd L. Slesinger, MD, FACEP, FCCM, FCCP, Program Director, Emergency Medicine, Aventura Hospital and Medical Center, FL.
Statement of Financial Disclosure
To reveal any potential bias in this publication, and in accordance with Accreditation Council for Continuing Medical Education guidelines, we disclose that Dr. Farel (CME question reviewer) owns stock in Johnson & Johnson. Dr. Stapczynski (editor) owns stock in Pfizer, Johnson & Johnson, AxoGen, Walgreens Boots Alliance Inc., and Bristol Myers Squibb. Ms. Mark’s (executive editor) spouse works for a company that creates advertising for Uroplasty. Dr. Schneider (editor), Dr. Martel (author), Dr. Ownbey (author), Dr. Simmons (author), Dr. Slesinger (peer reviewer), and Mr. Landenberger (editorial and continuing education director) report no financial relationships with companies related to the field of study covered by this CME activity.
The emergency department (ED) approach to a possible acute stroke used to be easy: ensure the blood pressure and glucose were not too high or low, computed tomography (CT) scan to exclude intracranial hemorrhage, and admit to hospital. Accuracy and rapidity of diagnosis in the ED were not necessarily priorities — alternatives and complications could be sorted out upstairs. Then, tissue plasminogen activator (tPA) for acute ischemic stroke came along. Now, both rapidity and accuracy of diagnosis are expected. As we are finding out, about a quarter of patients with symptoms apparently due to an acute ischemic stroke have another condition that mimics acute stroke. This issue of Emergency Medicine Reports is devoted to a discussion of these stroke mimics, providing guidance to their identification. The opposite of stroke mimics are stroke chameleons — in which acute strokes are misdiagnosed as other conditions. But that discussion is for another day.
— J. Stephan Stapczynski, MD, FACEP, Editor
Background and Introduction
Acute ischemic stroke (AIS) is a common and significant cause of mortality and morbidity in the United States, ranking fifth among all causes of death.1,2 It affects upwards of 800,000 people per year.3 Approximately 600,000 of these cases represent a first stroke,4 with the remainder representing recurrent strokes. Stroke is also a leading cause of permanent cognitive and function-limiting disability.5 Therefore, given the significant associated mortality and morbidity, the American Heart Association’s (AHA) Get with the Guidelines initiative emphasizes rapid recognition and potentially aggressive treatment of patients presenting with possible stroke symptoms.6,7
Prompt recognition of an AIS is crucial, as prior imaging-based work has demonstrated that the volume of irreversibly damaged brain tissue expands rapidly until reperfusion occurs.8 In a recent U.S. registry study, rapid recognition followed by early thrombolytic treatment was associated with improved outcomes. For every 15-minute faster interval to treatment, stroke patients experienced reduced mortality and reduced symptomatic intracranial hemorrhage (sICH), as well as increased rates of independent ambulatory status at time of discharge and discharge directly to home.9
However, timely recognition and treatment is complicated by the fact that there are multiple conditions that mimic acute ischemic stroke. A comprehensive review suggested that approximately 74% of patients presenting with apparent acute stroke symptoms were ultimately diagnosed with stroke,10 thus indicating that 26% of patients had their symptoms produced by “stroke mimics.” This estimation was similar to that from a 10-year study of more than 8000 possible stroke patients evaluated by stroke teams, which found that 30% of patients had a stroke mimic.11 More recent data reflect similar results, with one study focusing on telestroke cases reporting a stroke mimic rate of almost 25%,12 while another study of patients transferred from outlying hospitals with concern for acute ischemic stroke had a mimic rate of 17%, nearly all of whom received tPA.13 Therefore, prompt diagnosis is complicated by a multitude of stroke mimic etiologies, including structural intracranial abnormalities, infection, syncope, vertigo, seizure and migraine patterns, as well as underlying psychiatric causes and demyelinating diseases.
While the incidence of stroke mimics varies among the published studies, there are commonalities. Seizure, migraine, psychogenic disorders, and toxic/metabolic etiologies have been widely reported as among the most common non-vascular conditions that mimic stroke.14,15,16 Data from a single hospital identified that common stroke mimics included seizures, syncope, sepsis, and functional disorders/conversion disorders.17 (See Figure 1.) Another center reported that toxic/metabolic disorders and seizure etiologies were most common.14
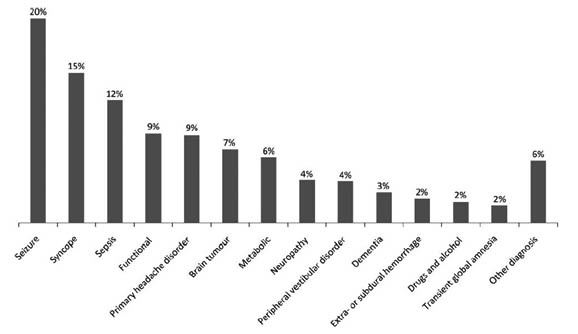
The wide array of potential stroke mimic etiologies often renders the clinical picture very difficult to distinguish from an acute ischemic process at the time of ED presentation. A factor that further complicates management is that patients may present within the appropriate thrombolysis administration window and subsequently receive this therapy. While it has been suggested that up to 14% of mimic patients receive thrombolytics,14,15,18 it has also been postulated that there is decreased risk of associated adverse effects in this population.14 There are no current data to suggest that tPA administration leads to adverse events in patients presenting with clinical features concerning for ischemic stroke but who are ultimately diagnosed with a stroke mimic.19,20
The key to avoiding unnecessary and potentially harmful therapy is improved understanding of the various mimic entities, as well as attention paid to important historical and physical exam features. In considering stroke symptoms, it can be helpful to think in terms of positive and negative symptoms. It has been explained elsewhere that positive symptoms are associated with excess central nervous system (CNS) activity, and are characterized by visual disturbance such as flashing lights and perception of various geometric shapes. In addition, somatosensory symptoms are associated with pain and/or paresthesias, and motor symptoms with jerking extremity movements. In contrast, negative symptoms are associated with a decrease in CNS function, and commonly manifest as acute vision or hearing loss, as well as sensorimotor deficits in the extremities.17 As a rule, acute ischemic stroke impairs CNS function manifested by negative symptoms. The three most common are unilateral subjective arm weakness, unilateral subjective leg weakness, and reported speech disturbance; positive symptoms are uncommon. Ischemia in the major cerebral vascular territories produces classic findings. (See Table 1.)
Territory |
Features |
|
Anterior cerebral |
Weakness and sensory loss, predominantly in the contralateral leg Aphasia Cognitive, behavioral, and emotional disorders may be prominent findings |
|
Middle cerebral (anterior portion involving motor cortex) |
Contralateral hemiparesis If involving dominant hemisphere: Broca’s (expressive or nonfluent) aphasia |
|
Middle cerebral (posterior portion involving sensory cortex) |
Contralateral hemineglect and sensory deficits Ipsilateral gaze preference Hemianopia If involving dominant hemisphere: receptive aphasia |
|
Lacunar (deep penetrating small arteries) — 5 classic subtypes |
1. Pure motor — contralateral weakness, facial weakness, ± dysarthria 2. Sensorimotor — contralateral hemiparesis, hemisensory loss 3. Pure sensory — contralateral sensory loss 4. Ataxic hemiparesis — homolateral ataxia and weakness 5. Dysarthria — clumsy hand — dysarthria, one hand clumsiness, ± central facial paralysis, dysphagia, tongue deviation |
|
Posterior cerebral |
Ipsilateral cranial nerve palsy with contralateral motor and/or sensory defect Loss of conjugate gaze Homonymous visual field defect |
Although it may be difficult to rapidly differentiate such a wide array of etiologies from ischemic stroke in the ED environment, it has been suggested that several key features are associated with a potential stroke mimic profile, including younger patients, milder symptoms, arrival to the ED by means other than emergency medical services (EMS), and lack of cardiovascular risk factors.11 Furthermore, several studies have suggested that patients with stroke mimics exhibit a lower prevalence of ischemic stroke risk factors.15,19 One large prospective study reported that specific historical factors associated with increased odds of having a stroke mimic included absence of hypertension, hyperlipidemia, and atrial fibrillation.11 This is a dilemma that emergency physicians will have to continually address as the treatment for acute stroke continues to evolve. The primary objective in this article is to review common stroke mimic etiologies, as well as discuss the key historical and physical exam characteristics associated with stroke mimics, and to present a diagnostic framework for approaching such cases.
Common Stroke Mimics: Seizure
Among stroke mimic diagnoses, seizure is one of the most common, being the final diagnosis in up to 20% of stroke mimics.21,22 For those patients with a prior stroke history, seizures may originate from previous ischemic or hemorrhagic stroke foci.17 The distinction is less complicated when the patient presents following a witnessed account of seizure activity and has a known history of epilepsy. A constellation of typical symptoms, including generalized convulsions, involuntary movements, postictal confusion, tongue-biting, and incontinence, make an acute ischemic event less likely.23 The clinical picture is clearly complicated by post-seizure paresis, and in one study this was the most frequent final diagnosis made among stroke mimics that received tPA.24 Although actual seizure activity is a relative tPA contraindication, more than 8% of patients in one study exhibited seizure activity after acute stroke symptom onset.25 Previous magnetic resonance imaging (MRI)-based studies have suggested that postictal dysfunction may relate to transient cerebral perfusion mismatch associated with epileptic foci but not with an ischemic lesion in a discrete vascular distribution, as would be seen in a stroke.26
Postictal paresis, or Todd’s paresis (or paralysis), may be difficult to distinguish from stroke. Reports vary with regard to the incidence and duration, with some studies suggesting that it occurs in as many as 10% of patients with generalized seizures, and may last for several hours or even longer in some cases.23 However, the vast majority of postictal deficits are commonly short, with one video monitoring study reporting a median time of 174 seconds in the case of focal epilepsy.27 Deficits may persist longer in the case of generalized seizure, with reports of postictal deficits lasting days. Importantly, focal motor deficits are not the only possible deficit following a seizure. Postictal dysphasia has also been reported to follow dominant hemisphere seizures.23 MRI with diffusion-weighted imaging (DWI) can assist in the differentiation between acute stroke and prior ischemic event, and may provide important information regarding the potential for post-stroke secondary seizure disorder.
Migraine
Migraine headache is a frequent ED complaint and leads to approximately 1 million patient visits annually.28 Migraine disproportionately affects females three times as often as males, with a peak incidence around age 40.28 Patients commonly present with a positive prior history and no evidence of focal neurological deficit on physical exam. However, in the setting of focal neurological deficit, primary headache disorders comprise up to 10% of stroke mimics.17 For instance, familial hemiplegic migraine is an autosomal dominant disorder with high penetrance. Since this presents with unilateral neurological deficits, it is particularly difficult to differentiate from acute stroke; however, there is a common pattern, as patients tend to be teenagers at the time of onset, and 70% are female.29 Likewise, migraine in general is thought to have a heritable component; if both parents have a history of migraine, there is a nearly 75% chance a patient will suffer from the disorder.30 It is characterized by recurrent severe headaches, classically of a unilateral and throbbing nature. Nausea, vomiting, photophobia, and phonophobia are common associated symptoms. Onset is generally gradual, and headaches may last up to three days.31 One particular diagnostic challenge is that nearly 30% of acute stroke patients report headache at the time of stroke symptom onset.32
Multiple theories have been proposed to explain the phenomenon of migraine headaches. The physiology of migraines is complex, with vasodilatory and inflammatory components. Although some alterations in local blood flow have been noted in patients experiencing migraines, the primary mechanism seems to be a triggered abnormality in the ability of the brain to manage sensory input.33 There are multiple subtypes that will be described in greater detail below.
Common migraines do not present with aura, and are the majority of cases. However, 20-30% of migraines are associated with aura, and may include a variety of neurological complaints and symptoms.33 These are known as classic migraines. It is these patients who are likely to raise suspicion for stroke when they present to the ED. By definition, migraine with aura is diagnosed after exclusion of organic disease, specifically TIA, ischemic stroke, and intracranial hemorrhage;31 in some patients with dense focal neurological deficits, this likely requires neuroimaging. Migraine auras generally exhibit a gradual onset over at least 5 minutes, and last a maximum of 60 minutes.33 The headache usually starts within one hour of the aura but may not always occur; it will lack the sudden/maximal onset or thunderclap component attributed to acute subarachnoid hemorrhage. Migraine auras can include a variety of neurological symptoms. Visual symptoms are most common, including scotomas, flashes/sparks of light, and zigzag-type visual phenomena.31
Although less common, paresthesias, motor disturbances, cognitive changes, and language deficits may also be seen in conjunction with aura. Ophthalmoplegic migraine includes involvement of cranial nerves III, IV, or VI with associated eye movement palsies and pupil changes. The third cranial nerve is most commonly involved, and the headache is ipsilateral to the deficits. Retinal migraine may result in scotoma or blindness. In the descriptively named hemiplegic migraine briefly described above, the aura includes motor symptoms such as hemiparesis or hemiplegia. In contrast to many ischemic stroke syndromes, the onset is typically gradual and associated with a visual, sensory, or speech disturbance. Basilar-type migraines present with an aura suggestive of brainstem vascular territory deficits and may exhibit multiple neurological findings, including visual loss (e.g., blindness), dysarthria, tinnitus, vertigo, bilateral paresthesias, paresis, and altered mentation.
In rare cases, an aura may become permanent. If this occurs, and an area of infarct is confirmed by neuroimaging in the distribution of the aura, it is termed a migrainous infarction or migrainous stroke.31 The pathophysiology of this phenomenon is unclear and remains, thankfully, rare. In the event that a patient with known migraine with aura experiences new or worsening aura symptoms or experiences aura that persists longer than normally would be expected (e.g., > 60 minutes), consider neurology consultation and advanced imaging.
Functional Disorders
Underlying psychiatric disease poses one of the most difficult challenges in the clinical diagnosis of acute stroke vs. stroke mimic, as patients may present with acute motor or sensory weakness in the setting of a specific emotional trigger.29,34 In particular, conversion disorder is a frequent cause of stroke mimic; in one study it was deemed the underlying etiology in nearly 75% of patients ultimately diagnosed with stroke mimic who received thrombolytics.13 Despite this finding, the prevalence of conversion disorder is rare overall, and it is essentially a diagnosis of exclusion not made in the acute care setting. Conversion disorder typically occurs more commonly in women, with the majority of cases seen in early childhood and adolescence. Complaints are widely varied, and may include paralysis, aphonia, seizures, ataxia/coordination difficulty, blindness, and anesthesia/paresthesia. Historically speaking, there may be a disproportionate lack of concern relative to the severity of symptoms (la belle indifference). With regard to physical exam, the hallmark is deficit inconsistency observed during serial physical exams, motor-sensory impairments with incongruent vascular territory distribution, and inconsistent task-dependent weakness or “give-away weakness.”17 However, this has also been observed in patients with concomitant organic disease and cannot be used to definitively diagnose conversion disorder.35 As such, the diagnosis is generally made only following admission and extensive neurological workup.
Functional disorders as a whole are thought to represent a physical manifestation of emotional distress,36 with common triggers, including panic attacks and dissociative episodes.34 Although providers may be tempted to directly confront patients in the acute setting, patients often lack insight into their condition, and the approach may consequently worsen the symptoms. In some cases, providing reassurance to the patient may rapidly improve or resolve symptoms, and if there is an acute stressor identified, then all efforts should be made to remove this from the equation.
Consider an underlying functional disorder if the symptoms are not consistent with a discrete vascular territory, if there are no objective neurological findings, and if there is an inconsistent physical exam. Unfortunately, there is no diagnostic study that can reliably provide this diagnosis. Patients with underlying psychiatric pathology also develop concomitant organic disease, and there are cases of patients being diagnosed with a functional disorder but ultimately found to have organic neurological pathology to explain their symptoms.35 Providers should be cautious in documenting a non-anatomic exam, atypical presentation, inconsistent physical exam, or a patient’s lack of concern about their symptoms as justification for not aggressively evaluating for acute stroke.
Systemic Infections
Sepsis may present with stroke-like features, and the most common associated infection is a urinary tract infection.37 Commonly associated symptoms include hyperthermia/hypothermia, tachycardia, and altered mental status, with or without an obvious historical or physical exam indication of an infectious source. However, the fact that fever may not be present and that a source etiology may not be immediately obvious during initial evaluation warrants consideration of an acute neurological insult as a potential cause of altered mental status.
In some cases, sepsis is a stroke risk factor, as severe sepsis has been shown to induce a hypercoagulable state.38 Furthermore, acute infection may result in recurrence of the focal neurological deficits initially present in patients who have a history of prior stroke, even when there is no residual deficit post-rehabilitation at baseline.39 Alternatively, there also may be cases of acute stroke with concomitant secondary infection, such as aspiration pneumonia.17
Obtaining a thorough past medical history, either from the patient or a family member, to establish baseline functional status is crucial but not always possible in the acute setting. When the history is unclear, objective focal neurological deficits should be assumed to stem from an acute neurological insult if history, vital signs, and physical exam do not clearly suggest an underlying infectious etiology. However, the timeframe of symptom onset may add some clarity to the evaluation. Sepsis is generally a gradual process and is less likely to present with the abrupt onset of acute, focal neurological deficits. In addition to more common sepsis sources, infections of the CNS, such as brain abscess, meningitis, and encephalitis, should also be considered in the setting of altered mental status.
Space-Occupying Lesions
Space-occupying brain lesions, including primary CNS tumors, metastases, and abscesses, have been shown to cause stroke-like focal neurological deficits attributed to alterations in cerebral blood flow associated with mass effect.40 However, mass effect is thought to progress slowly over a 24-48 hour period, and does not necessarily present with the sudden, acute change typically associated with an ischemic stroke process.41 Structural brain lesions are an infrequent cause of stroke mimic, and are subsequently less common than seizure, hypoglycemia, syncope, sepsis, functional disorders, and primary headache.42 Fortunately, noncontrast enhanced brain CT is readily available in almost all EDs and can assist in the rapid diagnosis of most acute space-occupying brain pathology, as well as acute intracranial hemorrhage.
Hypoglycemia
Transient hypoglycemia, easily assessed by bedside fingerstick glucose, commonly manifests with acute mental status changes and autonomic symptoms such as diaphoresis and tachycardia. However, it also has long been known to manifest with a constellation of stroke-like symptoms such as hemiplegia and aphasia in certain cases.43 Hypoglycemia-associated stroke-like presentations have been attributed to a wide variety of etiologies, including insulin or sulfonylurea medication use/overdose and alcohol consumption,44 as well as less common endocrine sources such as Addisonian crises and insulinomas.17 When rapidly recognized, hypoglycemia is easily correctable, and patients often quickly improve; however, there are reports of symptoms taking up to several hours to fully resolve.45 It is very important to address this prior to consideration of tPA administration, and national stroke guidelines mandate that a blood glucose level be obtained prior to tPA administration.7
Peripheral Neuropathy
Peripheral neuropathies are due to cellular level changes in nerves distal to the brain and spinal cord. In addition to potential toxic-metabolic, ischemic, or inflammatory involvement, the anatomic distribution of these structures relative to surrounding bony and soft-tissue structures places them at risk for acute compression and entrapment.24
Mononeuropathies may cause focal sensorimotor deficits, and in some instances may easily be mistaken for stroke symptoms when they occur in an acute timeframe. For instance, acute hand weakness may be seen in contralateral cortical strokes that resemble ulnar and radial nerve neuropathy. An acute peripheral nerve injury with subsequent myelin damage may be followed by Wallerian degeneration and result in sensorimotor dysfunction, muscle atrophy, and areflexia.24
The history and chronicity of symptoms will aid in distinguishing between acute neuropathy and stroke. However, symptoms associated with acute nerve compression and ischemia may exhibit similar chronicity, and subsequently are more likely to be confused. A high-quality physical exam in addition to detailed history is key to rapidly differentiating between these two entities. Table 2 details common peripheral neuropathy syndromes that can cause significant weakness and thus be confused with stroke.
Nerve Involved |
Mechanism/History |
Signs and Symptoms |
|
Radial nerve compression |
“Saturday night palsy” compression of the radial nerve in the spiral groove on the humerus, occurs from sleeping on the outstretched arm, prolonged compression over the back of a chair, etc. |
Wrist drop, finger extensor weakness, sensory loss on the dorsum of the hand |
|
Ulnar nerve compression |
Most commonly occurs at the elbow due to compression from leaning on a table or similar mechanism |
May cause weakness in the interosseous muscles, decreased grip, sensory loss of the fourth and fifth digits |
|
Cervical radiculopathy |
Nerve root compression at the neck |
Radicular pain is a key feature, specific distribution of weakness will depend on the nerve root C5: Shoulder abduction C6: Wrist extension C7: Wrist flexion C8: Finger flexion T1: Finger adduction and abduction |
|
Lumbosacral radiculopathy |
Spinal nerve root compression in the vertebral foramina, herniated disc |
Radicular pain is a key feature, deficits will depend on the nerve root L2-4: Hip flexion, knee extension L5: Foot dorsiflexion S1: Leg extension, foot plantar flexion |
|
Common peroneal nerve compression |
Compression of the peroneal nerve at the neck of the fibula, leg crossing, squatting, cast |
Foot drop, sensory loss on dorsum of foot and lateral shin |
|
Sciatic nerve compression |
Compression of the sciatic nerve at the sciatic notch, trauma, prolonged bed rest or seated position, piriformis syndrome |
Radicular pain to the back of the thigh and leg, weakness in the lower leg muscles, numbness sparing the medial calf |
One common mononeuropathy is radial nerve palsy, which is classically termed “Saturday night palsy.” This may occur with prolonged radial nerve compression (as with an arm draped over a chair) or with other prolonged periods of upper extremity immobilization such as during sleep. Compressive radial neuropathies typically occur distal to the branches that innervate the triceps muscle. Although a patient with radial neuropathy is likely to exhibit weakness with wrist and finger extension, arm extension is expected to remain intact, whereas a stroke patient might have concomitant triceps weakness in addition to the hand and wrist deficits. In addition, radial neuropathy sensory deficit localizes to the hand dorsum, while in acute stroke it is likely to be more volar in nature.46
Unilateral Facial Paralysis
Unilateral facial motor weakness is seen in both central and peripheral nerve pathologies. It involves upper motor neurons/contralateral motor cortex lesions or lower motor neurons, respectively. The two pathological foci may be distinguished by a notable difference in the distribution of motor weakness on physical exam. Peripheral lesions (e.g., Bell’s palsy) typically manifest as impairment in both upper and lower facial muscle function, whereas central lesions exhibit forehead sparing due to bilateral cortical innervation, therefore resulting in intact forehead wrinkling. In addition to differences in the distribution of central compared to peripheral facial paralysis, Bell’s palsy also may exhibit additional findings not typically seen in stroke, including decreased tear production (in some cases may also be excessive), acute gustatory impairment, and increased auditory acuity and facial hyperesthesia.47
Bell’s palsy is the most common cause of unilateral idiopathic facial palsy. It is often preceded by prodromal illness and is commonly attributed to herpes simplex virus infection.48 It has been more recently associated with Lyme disease in endemic regions. Lyme disease is the most common vector-mediated infection in the United States, is caused by the spirochete Borrelia burgdorferi, and is vectored by ticks in the genus Ixodes. Bell’s palsy represents the most common neurologic abnormality in the second stage of this important disease, and up to 25% of Bell’s palsy cases are attributed to Lyme disease.49 Furthermore, it is responsible for half of pediatric facial paralysis cases in endemic regions.50
Vertigo
Acute dizziness is a very common complaint in the ambulatory clinic setting, comprising up to 5% of visits. This represents nearly 10 million such visits annually, with 25% presenting in the ED setting.51 Acute dizziness is somewhat open to interpretation and is often described as vertigo, presyncope, and other unspecified unsteadiness in space that persists anywhere from minutes to hours, and is often accompanied by nausea, vomiting, nystagmus, and intolerance with head position change.52 Acute vestibular syndrome (AVS) presents with these symptoms, and is generally presumed to be a viral or post-viral phenomenon. Peripheral AVS generally occurs in the absence of focal neurologic deficits (such as acute unilateral motor and/or sensory deficits and gaze palsy) that would be associated with stroke.52
The diagnostic challenge posed in the acute setting is that 35% of posterior stroke cases present with dizziness as the chief complaint,53 whereas up to 3% of patients with acute dizziness have an ischemic stroke involving the posterior circulation. Ten percent of isolated posterior circulation stroke patients will present with isolated vertigo.54 However, the vast majority of patients presenting with dizziness are not having an acute stroke, and several groups have reported historical and diagnostic considerations for distinguishing these two entities clinically. Posterior stroke usually manifests with a constellation of neurological deficits and not isolated vertigo. Vertigo red flags suggesting a potential stroke etiology include any neurological exam deficit, total ipsilateral hearing loss, new ataxia with inability to walk without support, and direction-changing nystagmus.53 In particular, 71% of patients diagnosed with cerebellar stroke reported acute inability to walk without support.54 The vast majority of patients with serious pathology underlying their dizziness are older, have multiple cardiovascular risk factors, and have abnormal neurological exams in addition to dizziness, as up to 40% of posterior strokes are attributed to an embolic source.54
Thorough neurological examination also helps differentiate between these two disparate causes of dizziness. Ataxia requiring new ambulatory support and direction-changing nystagmus are particularly important in evaluating vertigo, since one of these two signs was seen in 84% of patients with cerebellar infarction and isolated vertigo.54 Furthermore, performing the Horizontal Head Impuse Test (h-HIT) also may help distinguish between peripheral and central etiologies specifically in the setting of vertigo. The h-HIT is a head acceleration maneuver performed on alert patients with active vertigo and designed to interrogate the vestibulo-occular reflex.55 It has been well described in the literature and essentially consists of examiner-directed random movement of the patient’s head. Patients visually fixate on the examiner’s eyes while the head is moved randomly from side to side. A positive test result indicates a peripheral lesion and manifests as a corrective saccade such that as the patient’s head is turned away, the patient’s eyes initially do not track the examiner but are followed by a rapid visual correction such that the eyes snap back to the original fixation point. Likewise, the lack of a corrective saccade raises suspicion for a central vertigo etiology.
The Head Impulse-Nystagmus Test of Skew (HiNTS), a variation of the h-HIT, approaches 100% sensitivity and 96% specificity for diagnosing posterior stroke when more than one component is abnormal.53 However, a considerable amount of training is required for diagnostic efficacy with this approach, and a combination of imaging and neurological consultation may be required to differentiate between peripheral and central etiologies.
Neurodegenerative Disorders
Acute demyelinating disorders, as well as initial diagnosis or acute exacerbation of multiple sclerosis (MS), may present with a wide variety of focal neurological deficits, including hemiparesis and cranial nerve or visual deficits.24 Episodes of acute diplopia, dysarthria, ataxia, and sensory deficits, often exacerabated by warm weather, are common with MS.56
Optic neuritis is an acute inflammatory disorder of the optic nerve that may present with sudden unilateral vision loss, and occurs more frequently in young adult women.57 It is a common initial manifestation of MS, occurring in 15-20% of these patients. In addition, up to half of MS patients may develop the condition at some point during their disease course.58
Acute disseminated encephalomyelitis (ADEM) is a post-infectious or post-immunization immune-mediated demyelinating disorder attributed to transient autoimmune destruction of myelin59 with multifocal brain lesions visible on MRI that generally occurs within three to six weeks of the inciting event.60 This time course, as well as a wide array of concomitant symptoms, including acute altered mental status, ataxia, optic neuritis, and sensorimotor deficits, aid in distinguishing this from an acute stroke syndrome.
Acute sensorimotor deficits also may be associated with non-compressive transient spinal cord inflammation. Although acute myelopathy is seen in the setting of ADEM,56 it may also manifest as transverse myelitis.59 This condition is generally not associated with acute back pain, and patients present with a wide variety of acute neurological deficits, including focal sensorimotor weakness and potential impairment of bowel, bladder, and sexual function below the lesion level.61,62
Similarly, Guillain-Barré syndrome is an acute inflammatory demyelinating polyneuropathy (AIDP) comprised of several subtypes. Although the less common of these varieties may be difficult to distinguish from acute stroke, it typically manifests as a progressive, ascending bilateral paralysis characterized by peripheral motor weakness, hyporeflexia, and, in advanced cases, respiratory distress when there is compromise of diaphragm innervation.24 In contrast, acute strokes typically manifest with unilateral deficits as previously described. However, brainstem infarcts in the basilar artery territory may present with a myriad of symptoms, including initial hemiparesis that may progress to bilateral limb weakness or locked-in syndrome, altered sensorium, vertigo, ataxia, bulbar motor weakness with gaze palsies or miotic pupils, as well as so-called “crossed-findings” with ipsilateral cranial nerve deficits and motor or sensory findings on the opposite side.63,64
Summary: Approach to Stroke Mimics
Having reviewed common stroke mimics, we can now consider how to approach evaluation of the patient with stroke-like symptoms. It is important to remember that acute stroke likely remains the most life-threatening and time-sensitive diagnosis for patients presenting to the ED with acute, focal neurological deficits. With few exceptions, the diagnosis of stroke mimic will only be made following appropriate resuscitation, laboratory evaluation, and advanced imaging, as well as neurology consultation.
The American Heart Association has developed guidelines for the initial assessment of suspected stroke,7 and stroke mimics are included in the list of alternative diagnoses to consider during the initial assessment. Recommended ancillary tests are basic blood tests, including serum glucose (rapid fingerstick is initially preferred for expedience), electrolytes, blood urea nitrogen/creatinine (BUN/Cr), complete blood count, troponin, and coagulation studies (PTT, PT/INR, possibly thrombin time [TT] or Ecarin Clotting Test [ECT] if taking direct thrombin inhibitors), as well as 12-lead electrocardiogram (ECG) and oxygen saturation. In most community hospitals, noncontrast enhanced CT scan of the brain is readily available and adequate to rapidly exclude intracranial hemorrhage in a patient eligible for tPA therapy.7 In specialized stroke centers, emergent MRI may be available to acutely map ischemic brain tissue and provide assistance for reperfusion therapy.
As with all patients, a thorough history and physical exam should be performed in a timely manner, including a baseline NIH stroke scale (NIHSS). Two or more of three key symptoms (acute facial paralysis, arm drift, or speech change) are associated with increased odds of having sustained an acute stroke (odds ratio [OR] 5.5; 95% CI: 3.3–9.1). Conversely, the absence of all three of these symptoms is thought to be helpful in identifying patients unlikely to have had an acute ischemic stroke.65 Particular historical factors have been associated with acute stroke diagnosis in multiple studies, and include older age, higher NIHSS, and a history of atrial fibrillation, hypertension, or diabetes. Likewise, patients who are young, have a low initial NIHSS, are initially unresponsive, have neurological exams incongruent with a anatomic vascular distribution, and patients with a history of epilepsy were more likely to be diagnosed with stroke mimics.11,13,21,22,39,66-69 One study reported that a history of psychiatric diagnosis was associated with greater likelihood of ultimate stroke mimic diagnosis.13
Some stroke mimics are more easily detectable. For instance, if hypoglycemia is identified, correct blood glucose levels and evaluate for functional response. If a space-occupying lesion is noted per noncontrast brain CT (ICH, tumor, shift, abscess), tPA is clearly contraindicated, and further diagnostics and treatment should be lesion-specific. If symptoms rapidly resolve and were preceded by a witnessed seizure or there is a history of epilepsy, consider Todd’s paralysis as a cause of focal neurological deficits. Although postictal exam findings in the potential unwitnessed seizure that support this diagnosis include tongue biting, bowel or urinary incontinence, and confusion, these objective findings may also be seen with acute stroke in some cases. In the event of a new focal neurological deficit, the initial workup remains unchanged, and if symptoms persist the patient likely warrants admission with neurological consultation. This type of presentation should be assumed to stem from an acute stroke until proven otherwise.
Specific attention should be given to episodes of prior neurological insult or known pathology, including stroke, seizures, migraine, and mass lesions. If available, a well-documented physical exam is very useful for comparison with a particular patient’s clinical picture at the time of presentation. For patients having sustained a prior stroke, all potential focal neurological deficits should be considered within the context of their medical history, past presenting symptoms, and subsequent residual deficits. Although this information may help provide context for the severity of a patient’s symptoms, it is important to carefully consider subjective components of the patient’s complaint as well as objective exam findings during surveillance for acute stroke pathology.
Table 3 includes a list of questions for providers to consider when a patient presentation is concerning for transient ischemic attack vs. a mimic.23 These criteria can also be applied to stroke, and may help to develop a differential diagnosis based on symptoms and time course. With any potential stroke patient, frequent re-evaluation during the workup is useful both to help clarify the extent of symptoms as well as to monitor for improvement or deterioration. Temporal changes in symptoms over time may gradually move the level of suspicion closer to or farther from acute stroke.
|
Age and other demographic data: Is there a high a priori probability of a cerebrovascular event? |
As noted above, certain demographic and historical factors have been shown to correlate with the likelihood of an event representing a stroke vs. a stroke mimic. |
|
Nature of the symptoms: Positive vs. negative |
Stroke is more likely to present with negative symptoms, whereas mimic due to migraine aura, conversion disorder, or other causes is more likely to have positive symptoms. |
|
Onset and progression |
Sepsis may be a slower onset, while seizure, stroke, and even tumor may present suddenly. |
|
Duration |
Rapidly resolving symptoms raise the question of a TIA or seizure. |
|
Precipitating factors |
Deficits preceded by a seizure or illness may indicate an underlying diagnosis. |
|
Associated symptoms, for example, headache, loss of awareness, during or after the attacks |
Headache may be associated with stroke but also with migraine. Loss of awareness may increase suspicion of seizure. |
|
Adapted with permission from: Nadarajan V, Perry RJ, Johnson J, et al. Transient ischaemic attacks: Mimics and chameleons. Pract Neurol 2014;14:23–31. |
|
Nevertheless, ED providers are in a unique position to rapidly identify acute neurological pathology, and it is incumbent on the front-line physician to consider acute stroke first. However, given the wide array of clinical mimics that complicate this process, a detailed understanding of such peripheral entities will enable rapid diagnosis of easily correctable entities (e.g., hypoglycemia) and will expediently resolve exactly which patients are appropriate for acute treatment (e.g., tPA) or require immediate subspecialist support (e.g., neurosurgery) from those for whom a diagnosis of exclusion to explain their symptoms will likely occur during hospitalization.
REFERENCES
- Centers for Disease Control and Prevention, National Center for Health Statistics. Compressed Mortality File 1999-2009. CDC Wonder Online Database, Compiled for Compressed Mortality File 1999–2009 Series 20, No. 20, 2012. Underlying Cause-of-Death 1999–2009. wonder.cdc.gov/Mortsql.Html. Accessed June 1, 2015.
- Kochanek KD, Xu JQ, Murphy SL, et al. Mortality in the United States, 2013. NCHS Data Brief, No. 178. Hyattsville, MD: National Center for Health Statistics, Centers for Disease Control and Prevention, US Dept. of Health and Human Services; 2014.
- Mozzafarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics — 2015 update: A report from the American Heart Association. Circulation 2015;131:e29–322.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics — 2014 update: A report from the American Heart Association. Circulation 2014;129:e28-e292.
- Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults — United States, 2005. MMWR Morb Mortal Wkly Rep 2009;58:421-426.
- Goldstein LB, Bushnell CD, Adams RJ, et al. Guidelines for the primary prevention of stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2010;42:517–584.
- Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013;44:870-947.
- Saver JL. Time is brain — quantified. Stroke 2006;37:263-266.
- Saver JL, Fonarow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013;309:2480-2488.
- Goldstein LB, Simel DL. Is this patient having a stroke? JAMA 2005;293:2391–2402.
- Merino JG, Luby M, Benson RT, et al. Predictors of acute stroke mimics in 8187 patients referred to a stroke service. J Stroke Cerebrovasc Dis 2013;22:397-403.
- Ali SF, Viswanathan A, Singhal AB, et al. The telestroke mimic (TM)-Score: A prediction rule for identifying stroke mimics evaluated in a telestroke network. J Am Heart Assoc 2014 Jun 23;3:e000838.
- Mehta S, Vora N, Edgell RC, et al. Stroke mimics under the Drip-and-Ship Paradigm. J Stroke Cerebrovasc Dis 2014;23:844-849.
- Brunser AM, Illanes S, Lavados PM, et al. Exclusion criteria for intravenous thrombolysis in stroke mimics: An observational study. J Stroke Cerebrovasc Dis 2013;22:1140-1145.
-
Chernyshev OY, Martin-Schild S, Albright KC, et al. Safety of tPA in stroke mimics and neuroimaging-negative cerebral ischemia. Neurology 2010;74:
1340–1345. - Hand PJ, Kwan J, Lindley RI, et al. Distinguishing between stroke and minimc at the bedside: The Brain Attack Study. Stroke 2006;37:769-775.
- Fernandes PM, Whiteley WN, Hart SR, et al. Strokes: Mimics and chameleons. Pract Neurol 2013;13:21–28.
- Hess DC, Audebert HJ. The history and future of telestroke. Nat Rev Neurol 2013;9:340-350.
- Scott PA, Silbergleit R. Misdiagnosis of stroke in tissue plasminogen activator-treated patients: Characteristics and outcomes. Ann Emerg Med 2003;42:611-618.
- Zinkstok SM, Engelter ST, Gensicke H, et al. Safety of thrombolysis in stroke mimics: Results from a multicenter cohort study. Stroke 2013;44:1080–1084.
-
Forster A, Griebe M, Wolf ME, et al. How to identify stroke mimics in patients eligible for intravenous thrombolysis?
J Neurol 2012;259:1247-1253. - Tsivgoulis G, Alexandrov AV, Chang J, et al. Safety and outcomes of intravenous thrombolysis in stroke mimics. Stroke 2011;42:1771-1774.
- Nadarajan V, Perry RJ, Johnson J, et al. Transient ischaemic attacks: Mimics and chameleons. Pract Neurol 2014;14:23–31.
-
Magauran BG, Nitka M. Stroke mimics. Emerg Med Clin North Am 2012;30:
795-804. -
Bladin CF, Alexandrov AV, Bellavance A, et al. Seizures after stroke: A prospective multi-centre study. Arch Neurol 2000;57:
1617-1622. - Rupprecht S, Schwab M, Fitzek C, et al. Hemispheric hypoperfusion in postictal paresis mimics early brain ischemia. Epilepsy Res 2010;89:355–359.
- Gallmetzer P, Leutmezer F, Serles W, et al. Postical paresis in focal epipelsies-Incidence, duration, and causes. Neurology 2004;62:2160-2164.
- Vinson DR. Treatment patterns of isolated benign headache in US emergency departments. Ann Emerg Med 2002;39:215-222.
- Thomsen LL, Eriksen MK, Roemer, SF et al. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain 2002;125:1379-1391.
- MacGregor EA. In the clinic: Migraine. Ann Intern Med 2013;159:ITC5-1–ITC5-15.
- Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd ed. Cephalalgia 2013;33:629-808.
- Tentschert S, Wimmer R, Greisenegger S, et al. Headache at stroke onset in 2196 patients with ischemic stroke or transient ischemic attack. Stroke 2005;36:e1-3.
- Kwiatkowski T, Friedman BW. Headache disorders. In: Marx J ed. Rosen’s Emergency Medicine: Concepts and Clinical Practice, 8th edition. Elsevier Health Sciences; 2014.
- Stone J, Warlow C, Sharpe M. Functional weakness: Clues to mechanism from the nature of onset. J Neurol Neurosurg Psychiatry 2012;83:67-69.
- Winter AO. Somatoform Disorders. In: Marx J, ed. Rosen’s Emergency Medicine: Concepts and Clinical Practice, 8th edition. Elsevier Health Sciences; 2014.
- Feinstein F. Conversion disorder: Advances in our understanding. CMAJ 2011;183:915-920.
- Tuntiyatorn L, Saksornchai P, Tunlayadechanont S. Identification of stroke mimics among clinically diagnosed acute strokes. J Med Assoc Thai 2013;96:1191-1198.
- Valtonen VV. Infection as a risk factor for infarction and atherosclerosis. Ann Med 1991;23:539-543.
- Hemmen TM, Meyer BC, McClean TL, et al. Identification of nonischemic stroke mimics among 411 code strokes at the University of California, San Diego, Stroke Center. J Stroke Cerebrovasc Dis 2008;18:23-25.
- Ueno Y, Tanaka A, Nakayama Y. Transient neurological deficits simulating transient ischemic attacks in a patient with 34 meningioma — case report. Neurol Med Chir (Tokyo) 1998;38:661–665.
- Hacke W, Schwab S, Horn M, et al. ‘Malignant’ middle cerebral artery territory infarction: Clinical course and prognostic signs. Arch Neurol 1996;53:309-315.
- Morgenstern LB, Frankowski RF. Brain tumor masquerading as stroke. J Neurooncol 1999;44:47–52.
- Montgomery BM, Pinner CA, Newberry SC. Transient hypoglycemic hemiplegia. Arch Int Med 1964;114:680-684.
- Andrade R, Mathew V, Morgenstern MJ, et al. Hypoglycemic hemiplegic syndrome. Ann Emerg Med 1984;13:529-531.
- Wallis WE, Donaldson I, Scott RS, et al. Hypoglycemia masquerading as cerebrovascular disease (hypoglycemic hemiplegia). Ann Neurol 1985;18:510-512.
- Tarulli A. Neurology: A Clinician’s Approach. Cambridge University Press, 2010: 91.
- Marenda S, Olsson J. The evaluation of facial paralysis. Otolaryngol Clin North Am 1997;30:669.
- Murakami S, Mizobuchi M, Nakashiro Y, et al. Bell palsy and herpes simplex virus: identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med 1996;124:27-30.
- Ho K, Melanson M, Desai JA. Bell palsy in Lyme disease-endemic regions of Canada: A cautionary case of occult bilateral peripheral facial nerve palsy due to Lyme disease. Can J Emerg Med 2012;14:321-324.
- Cook SP, Macartney KK, Rose CD, et al. Lyme disease and seventh nerve paralysis in children. Am J Otolaryngol 1997;18:320.
- Newman-Toker DE, Hsieh YH, Camargo CA Jr, et al. Spectrum of dizziness visits to US emergency departments: Cross-sectional analysis from a nationally representative sample. Mayo Clin Proc 2008;83:765-775.
- Tarnutzer AA, Berkowitz AL, Robinson KA, et al. Does my dizzy patient have a stroke? A systematic review of bedside diagnosis in acute vestibular syndrome. CMAJ 2011;183:E571–E592.
- Kattah, JC, Talkad AV, Wang DZ, et al. HINTS to diagnose stroke in the acute vestibular syndrome: Three-step bedside oculomotor examination more sensitive than early MRI diffusion-weighted imaging. Stroke 2009;40:3504–3510.
- Lee H, Sohn SI, Cho YW, et al. Cerebellar infarction presenting isolated vertigo: Frequency and vascular topographical patterns. Neurology 2006;67:1178-1183.
- Halmagyi GM, Curthoys IS. A clinical sign of canal paresis Arch Neurol 1988;45:737-739.
- Li Y, Zeng C, Luo T. Paroxysmal dysarthria and ataxia in multiple sclerosis and corresponding magnetic resonance imaging findings. J Neurol 2011;258:273–276.
- The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: Final optic neuritis treatment trial follow-up. Arch Neurol 2008;65:727–732.
- Marcel C, Anheim M, Flamand-Rouviere C, et al. Symptomatic paroxysmal dysarthria-ataxia in demyelinating diseases. J Neurol 2010;257:1369–1372.
- Tenembaum S, Chitnis T, Ness J, et al. Acute disseminated encephalomyelitis. Neurology 2007;68 (16 Supple 2):S23-36.
- Alexander M, Murthy JMK. Acute disseminated encephalomyelitis: Treatment guidelines. Ann Indian Acad Neurol 2011;14(Suppl 1):S60-S64.
- Beh SC, Greenberg BM, Frohman T, et al. Transverse myelitis. Neurol Clin 2013;31:79-138.
- Frohman EM, Wingerchuk DM. Clinical practice. Transverse myelitis. N Engl J Med 2010;363:564–572.
- Ferbert A, Brückmann H, Drummen R. Clinical features of proven basilar artery occlusion. Stroke 1990;21:1135–1142.
- Nouh A, Remke J, Ruland S. Ischemic posterior circulation stroke: A review of anatomy, clinical presentations, diagnosis, and current management. Front Neurol 2014;5:30.
- Gibson LM, Whiteley W. The differential diagnosis of suspected stroke: A systematic review. J R Coll Physicians Edinb 2013;43:114–118.
- Artto V, Putaala J, Strbian D, et al. Stroke mimics and intravenous thrombolysis. Ann Emerg Med 2012;59:27-32.
- Vroomen PC, Buddingh MK, Luijckx GJ, et al. The incidence of stroke mimics among stroke admissions in relation to age group. J Stroke Cerebrovasc Dis 2008;17:418-422.
- Chang J, Teleb M, Yang JP, et al. A model to prevent fibrinolysis in patients with stroke. J Stroke Cerebrovasc Dis 2012;21:839-843.
- Valle J, Lopera E, Guillan M, et al. Stroke mimics: A challenge for the emergency physician. An Sist Sanit Navar 2014;37:117-128.
MONOGRAPH: A quarter of patients with symptoms apparently due to an acute ischemic stroke have another condition that mimics it.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.