Mimics of ST Elevation Myocardial Infarction (STEMI)
AUTHORS
Larissa I. Velez, MD, FACEP, Michael P. Wainscott Professorship in Emergency Medicine, University of Texas Southwestern, Dallas.
Maxwell Hockstein, MD, Emergency Medicine Resident, Parkland Hospital, Dallas.
Jakub Furmaga, MD, Assistant Professor, Emergency Medicine, University of Texas Southwestern, Dallas.
Stephen W. Smith, MD, Faculty, Emergency Medicine Residency, Hennepin County Medical Center, Minneapolis, MN; Professor, Emergency Medicine, University of Minnesota.
PEER REVIEWER
William J. Brady, MD, FACEP, FAAEM, Professor of Emergency Medicine and Medicine, Medical Director, Emergency Preparedness and Response, University of Virginia Operational Medicine Director, Albemarle County Fire Rescue, Charlottesville, VA.
To reveal any potential bias in this publication, and in accordance with Accreditation Council for Continuing Medical Education guidelines, we disclose that Dr. Farel (CME question reviewer) owns stock in Johnson & Johnson. Dr. Stapczynski (editor) owns stock in Pfizer, Johnson & Johnson, Walgreens Boots Alliance Inc., GlaxoSmithKline, Bristol Myers Squibb, and AxoGen. Dr. Schneider (editor), Ms. Fessler (nurse planner), Dr. Velez (author), Dr. Hockstein (author), Dr. Furmaga (author), Dr. Smith (author), Dr. Brady (peer reviewer), Ms. Mark (executive editor), Ms. Coplin (executive editor), and Mr. Landenberger (editorial and continuing education director) report no financial relationships with companies related to the field of study covered by this CME/CE activity.
Early in my training, I remember reading an article that concluded 25% of myocardial infarctions (MIs) were missed. I know that we physicians are not perfect, but are we that bad? We miss a quarter of patients with an acute infarction who present to the ED? When I delved into this literature about missed MIs, I found that this “miss” rate was being determined by analysis that had nothing to do with ED patients. What were being determined were silent infarctions — those that did not produce enough clinical symptoms to prompt patients to come to the ED. Interestingly, a May 16 publication in Circulation found that of all MIs observed in about 9,400 individuals initially free of cardiovascular disease and followed for almost nine years, more than 45% were silent.
When I reviewed the ED literature on missed MIs, the observed miss rate varied between 2% and 4%. Thankfully, not a quarter, but also not zero. There were multiple factors associated with missing an MI, including patient and physician factors. Two ancillary testing factors were also associated with missing an infarction: the insensitive cardiac biomarker tests available in that era and physician misinterpretation of the initial ECG. I believe the miss rate has decreased further with the widespread adoption of sensitive and specific cardiac biomarkers used with a serial testing protocol. One factor that still bedevils emergency physicians is the appearance and potential for misinterpretation of the initial ECG.
The opposite of a missed infarction is a false-positive overcall of infarction, primarily due to disorders that can mimic an acute infarction by producing ST segment elevation. This issue of Emergency Medicine Reports discusses several of these conditions that are important for the emergency physician to recognize to appropriately manage patients in the ED and to better distinguish acute infarction from other conditions on the ECG.
— J. Stephan Stapczynski, MD, FACEP, Editor
Introduction
One common axiom in emergency medicine is that ST segment elevation in an emergency department (ED) patient with chest pain should be assumed to be an acute myocardial infarction (AMI) until proven otherwise. Like many axioms in emergency medicine, it embodies an “assume the worst and rule it out” approach to evaluating acute symptoms. While such a principle concerning ST segment elevation may be cautious and prudent, it is only one factor to consider when assessing patients with chest pain. The physician is also aware of the patient’s history, physical exam findings, and other features of the electrocardiogram (ECG) when determining the potential cause of ST segment elevation. In fact, the majority of ST segment elevation seen in ED patients with chest pain is not due to AMI.1 Thus it is important for emergency physicians to have an understanding of the differential diagnosis of ST segment elevation.
In 2013, the American College of Cardiology Foundation and the American Heart Association (ACCF/AHA) revised the electrocardiographic definition of ST elevation myocardial infarction (STEMI) to: “new ST elevation at the J point in at least 2 contiguous leads of ≥ 2 mm (0.2 mV) in men (≥ 2.5 mm in men under 40 years old) or ≥ 1.5 mm (0.15 mV) in women in leads V2–V3 and/or of ≥ 1 mm (0.1 mV) in other contiguous chest leads or the limb leads.”2 In the updated guidelines, a presumably new left bundle branch block (LBBB) in isolation is no longer considered STEMI equivalent. Moreover, the American College of Cardiology (ACC) emphasized that AMI is a syndrome, a constellation of clinical findings, including but not limited to findings on the 12-lead ECG that are concerning for an acute infarct, but also including the subsequent release of biomarkers indicative of myocardial necrosis.
The mechanism by which ST segment elevation occurs in an AMI is not completely elucidated;3 however, it is clear that ST elevation occurs reliably with transmural and subepicardial myocardial infarctions. In a classic study conducted in 1960, ST segment elevation was described as an “injury current,”4 after observing its presence in a canine myocardium after ligating its supplying coronary artery. In this experiment, the injured myocardium displayed simultaneous areas of depolarized and repolarized tissue, which resulted in ST elevation. A competing theory suggests that the surface of injured myocytes becomes more negative, inducing a positive charge in the surrounding (uninjured) myocytes, which produces elevation.5 Regardless of the mechanism, the final common pathway for all theories of why ST elevation occurs is due to an irregularity in repolarization.
There are many conditions that alter repolarization, and therefore can produce ST elevation, but which are not due to an acute MI. The purpose of this article is to discuss several disorders that can produce ST segment elevation and mimic a STEMI. These conditions can be remembered through the use of the “ELEVATION” mnemonic: Early repolarization, Left bundle branch block, Electrolytes, Ventricular enlargement, Aneurysm, Thailand (representative of South Asia, where Brugada syndrome has the highest reported incidence), Inflammation (myopericarditis), and Non-thrombotic vasospasm.
Early Repolarization
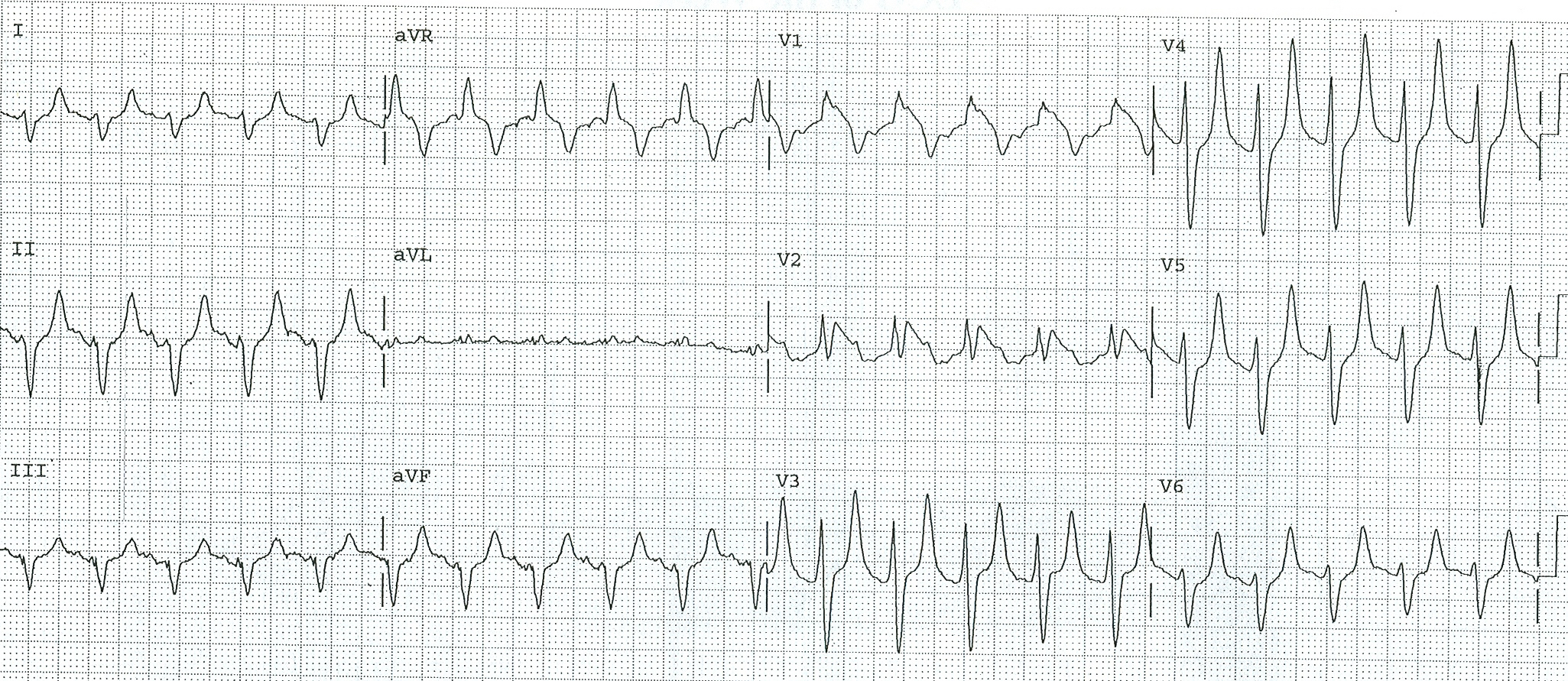
Historically, ECGs with early repolarization patterns have been regarded as a benign variant associated with persistent ST segment elevation in individuals with no evidence of cardiac disease. (See Figure 1.) In addition to ST segment elevation, slurring or notching on the downstroke of a dominant R wave is common. Early repolarization was initially associated with young healthy athletes,6 but is increasingly found in a wider variety of individuals. The dramatic appearance of ST segment in multiple contiguous leads results in early repolarization being cited as the most common caused of false-positive catheterization laboratory activations in patients without elevated cardiac biomarkers.7 Over the years, different authors have used different criteria for the diagnosis of early repolarization. In an effort to provide consistency, in 2015, Hancock et al proposed three criteria8 that are required for the diagnosis of benign early repolarization:
- The QRS slur or notch (termed a J wave) must be on the downslope of the R wave and be above the isoelectric line;
- The peak of the J point must be elevated ≥ 0.1 mV in two or more contiguous leads except V1-V3;
- The QRS duration must be < 120 ms.
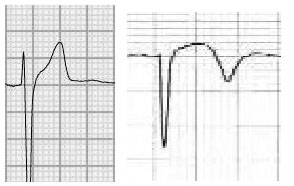
The above definition helps clinicians differentiate a normal electrocardiographic variant from ischemic ST elevation. In benign early repolarization, the ST elevation, if present, is described as concave (Figure 2, left), in contrast to a STEMI, which typically has a convex (Figure 2, right) ST elevation morphology. However, the ST segment convexity only confers a 77% sensitivity for infarction9 and, therefore, should not be used as the sole discriminating finding between early repolarization and infarction. Smith et al published predictors that help differentiate between subtle anterior wall STEMI over benign early repolarization. These include low R wave amplitude (best measured in V4), greater degrees of ST segment elevation, and longer computer-measured QTc.10 These criteria (available on Dr. Smith’s ECG blog11) are relatively complex to implement in a clinical setting; however, they offer highly sensitive (86%) and specific (90%) discrimination between anterior wall STEMI and early repolarization.
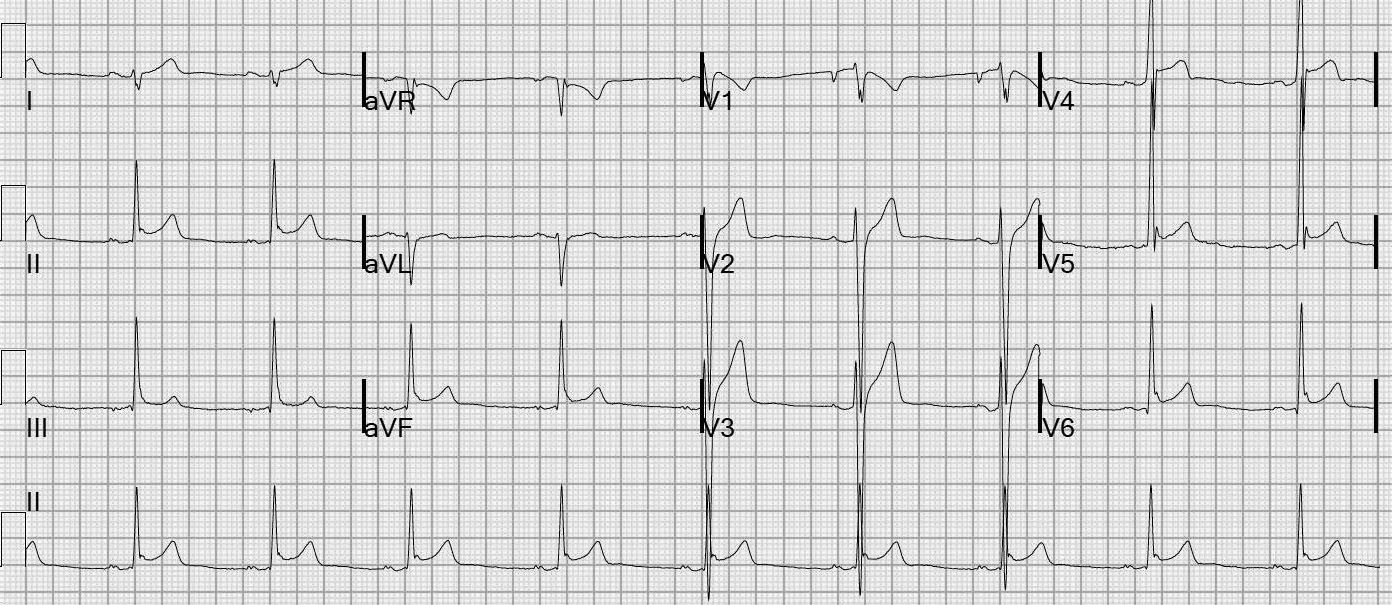
Electrophysiology literature suggests that early repolarization is associated, in some cases, with sudden cardiac death (idiopathic ventricular fibrillation), earning the term “early repolarization syndrome” (ERS) or “J wave syndrome.”12 The J waves seen in ERS may be indistinguishable from those that are true Osborn waves in hypothermia, as the underlying mechanism is identical. In addition, because ERS and Brugada syndrome (covered later in this article) are believed to be part of the same continuum, it is not surprising that both have been associated with polymorphic ventricular tachycardia and ventricular fibrillation (VF).12 While the absolute risk of sudden death with ERS remains unclear, it is estimated to be small (lower than that of Brugada syndrome). It is important to differentiate early repolarization pattern from ERS; the former is an ECG variant seen in asymptomatic individuals, whereas ERS is applied only after a documented VF arrest.
Left Bundle Branch Block
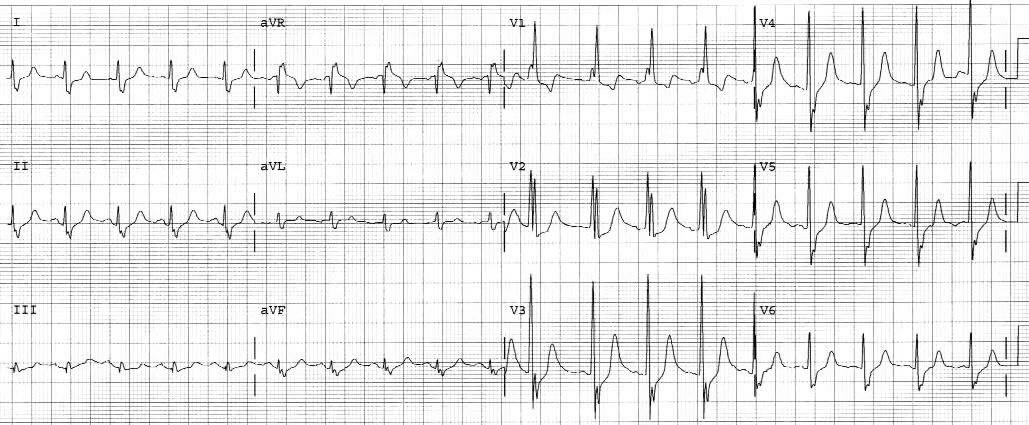
Left bundle branch blocks (LBBBs) are characterized by a QRS duration > 120 ms with features suggestive of depolarization from the right to the left ventricle: a dominant Q or predominant S in V1-V3 (QRS will be net negative) and a broad, dominant R wave in the lateral leads (I, aVL, V5, V6). (See Figure 3.) This QRS configuration is present in LBBB, but a variant is also encountered in a right ventricular paced pattern (as both of these cause ventricular depolarization to happen from right to left). A new LBBB was once regarded as a STEMI equivalent; however, after observing a relatively low frequency of acutely obstructing coronary lesions in ED patients with chest pain and a new LBBB, that recommendation was removed from the 2013 ACCF/AHA STEMI Management Guidelines.2 In hemodynamically stable patients with a presumed new LBBB, evaluation of their symptoms requires both measurement of cardiac biomarkers and observation. In patients with hemodynamic compromise (including acute heart failure), revascularization should be emergently considered.13
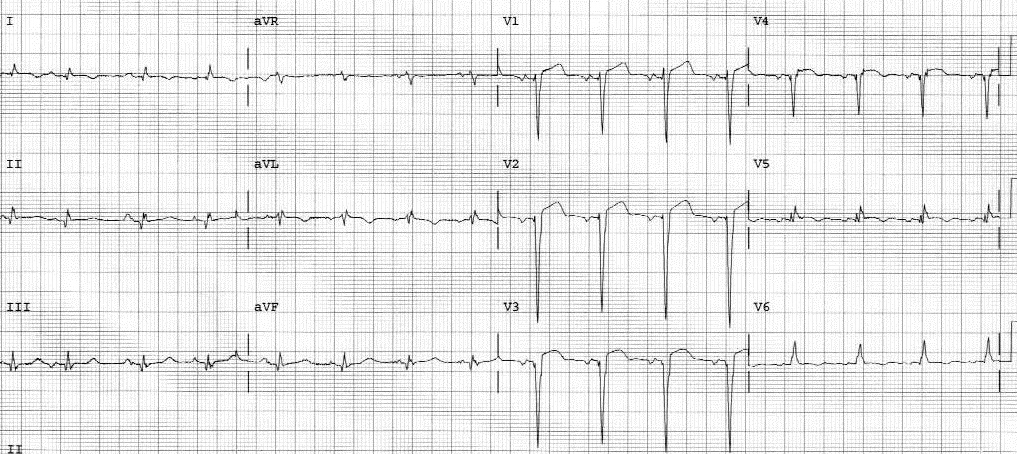
The Sgarbossa criteria14 can help guide the decision for emergent catheterization and coronary intervention in the presence of both new and old LBBBs. The normal state of LBBB is described by the “rule of appropriate discordance.”15 This idea is that ventricular repolarization (ST-T) occurs in the opposite direction of the majority of the ventricular depolarization (QRS), which manifests itself as the net polarity of the QRS and T wave being opposite from each other. Thus an ST segment in the same direction as the QRS (also known as “concordant“) is indicative of ischemia/infarction. Conversely, in LBBB, the QRS in V1-V3 is always negative; therefore, the normal condition of the ST segment in these leads is ST elevation. Thus, excessive discordant ST segment elevation in leads V1-V3 is indicative of an anterior MI. The modified Sgarbossa criteria16 determine “excessive discordance” by a proportion rather than an absolute number (these criteria have been validated18):
- Concordant ST elevation ≥ 1 mm in any single lead (see Figure 4);
- Concordant ST depression ≥ 1 mm in just one of leads V1-V3;
- Proportionally excessive discordant ST elevation as defined by a ratio of ST elevation at the J-point, relative to the depth of the S wave (ST/S ratio), of ≥ 0.25 (this has replaced the original third criterion of ST elevation, which was an absolute number [≥ 5 mm]). (See Figure 4.)
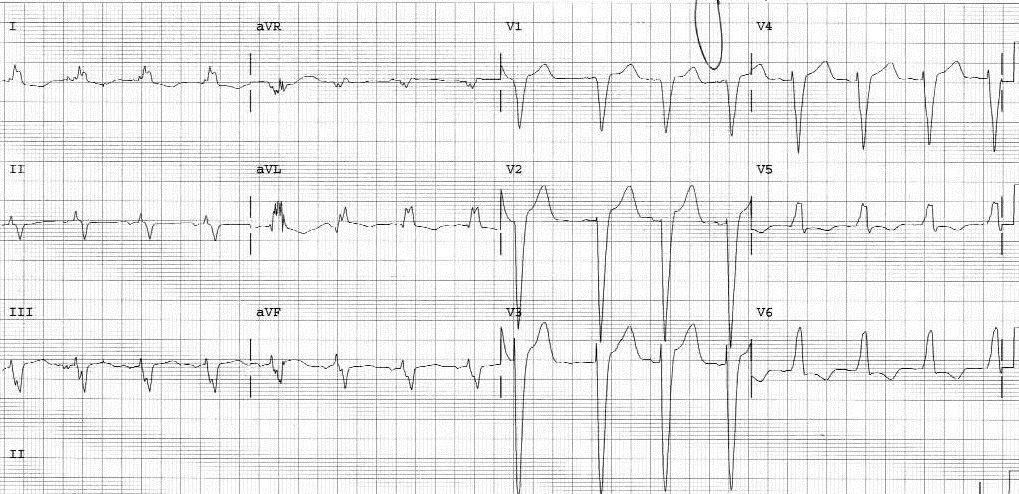
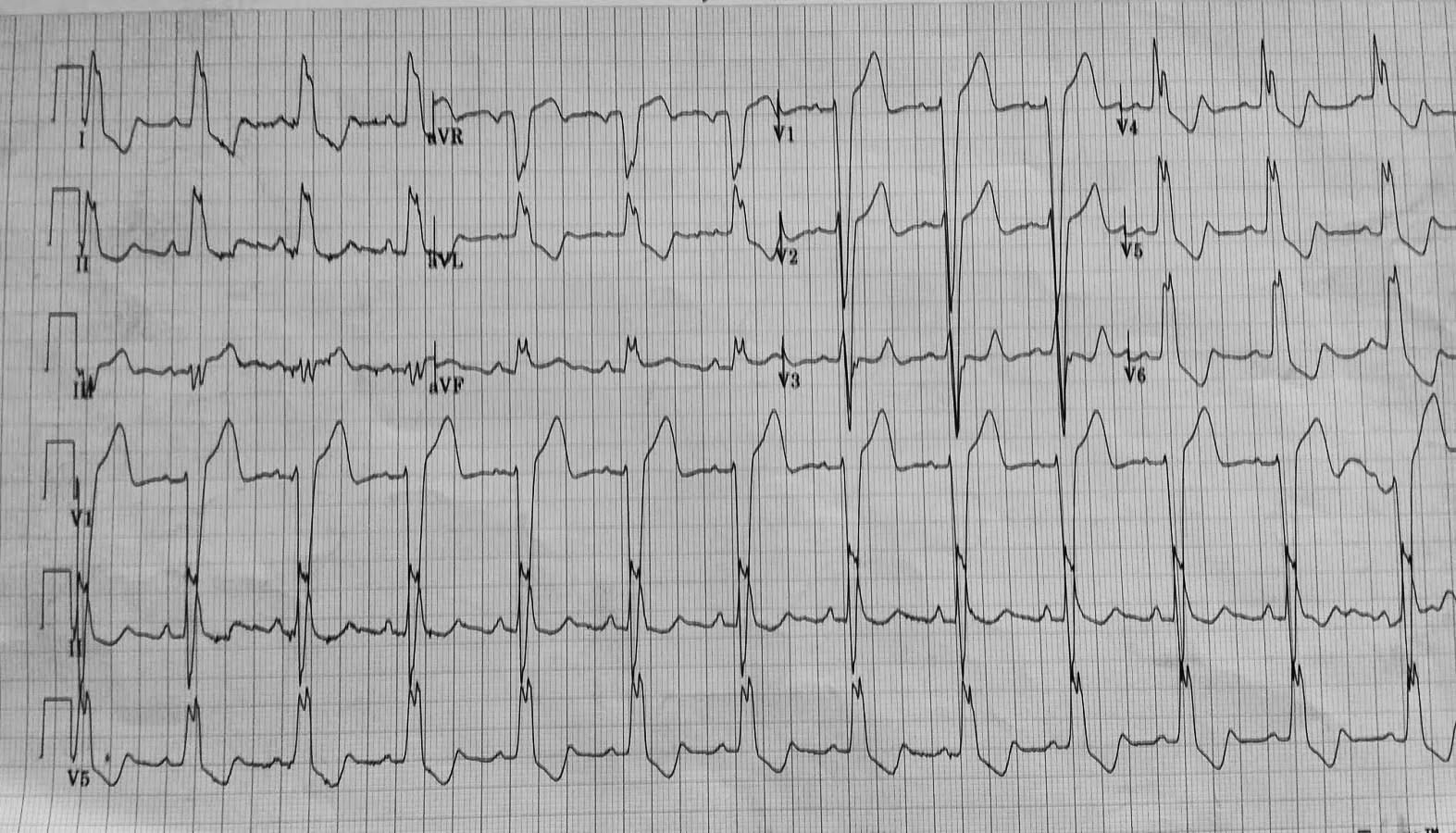
Image courtesy of Stephen Smith, MD.
The original Sgarbossa decision tool assigned points to each criterion: 5 points for concordant ST elevation > 1 mm in any lead; 3 points for concordant ST depression > 1 mm in leads V1 to V3; and 2 points for discordant ST elevation > 5 mm in any lead. With a threshold of 3 or more points, the initial article describing Sgarbossa criteria reported a sensitivity of about 80% and a specificity of about 90% for detecting AMI in the presence of a LBBB.14
In subsequent studies, other authors reported lower sensitivities, and a 2008 meta-analysis of 11 studies reported a summary sensitivity of only 20% and a summary specificity of 98% using a score of 3 or more.17 To increase sensitivity, the modified criteria have been proposed, and although they have not been subjected to extensive validation, the reported sensitivity from a few studies is about 80%.18 Also observed in the setting of LBBB, Chapman’s sign (a notching of the R wave seen in leads I, aVL, and sometimes V6) is occasionally seen in anterior wall MIs, as well as the analogous Cabrera’s sign (a notch in the S wave, seen mostly in V3 and V4). Both of these signs, although easier to appreciate than the Sgarbossa criteria, are less sensitive. These signs are Q wave equivalents and, therefore, are only indicative of a completed infarct and do not indicate ischemic or viable myocardium. The sensitivity and specificity of the Sgarbossa criteria as they pertain to patients with paced rhythms are still being investigated.
Electrolytes
Electrolyte derangements can cause almost any ST segment change. Changes in the concentrations of potassium, calcium, magnesium, and sodium alter the cardiac action potential, resulting in ECG changes.
Hyperkalemia frequently results in ST elevation, especially in leads V1 and V2, but occasionally in other leads. (See Figure 5.) It can be suspected when there is any QRS widening, especially when similar to a right bundle branch block (RBBB), and especially when associated with some symmetric peaking of the T waves (“T waves that will poke you if you touch them”). Other ECG changes in hyperkalemia include shortening of the QT interval, shortening of the PR interval, flattening of the P waves, loss of sinoatrial conduction resulting in a wide-complex (“sine-wave” or “sinoventricular”) rhythm, and, ultimately, ventricular fibrillation and asystole. The ECG changes might not occur in a stepwise fashion and are more dependent on the rate of potassium elevation than on the absolute value.19 Obviously, the best way to differentiate STEMI from hyperkalemia is to measure serum potassium. If the level is very elevated, correct it and repeat the ECG. The ECG findings should improve. Occasionally, they do not resolve because the ST elevation was indeed due to a simultaneous STEMI.
Calcium plays a key role in phase 2 (the plateau phase) of the cardiac action potential. It maintains a balance between inward calcium flow through the L-type calcium channel, coupled with outward potassium flow through the delayed rectifier potassium channel. This balance is affected by the presence of excess serum calcium. When present, hypercalcemia (see Figure 6) slows ventricular conduction velocity and shortens the refractory period of myocytes. Characteristic ECG changes of hypercalcemia include shortening of the QT interval and occasionally prolongation of the PR interval. This shortening of the QT interval is what may mimic ST elevation. Hypercalcemia may also cause the appearance of J waves (discussed later). Hypercalcemia can also be arrhythmogenic (including atrioventricular blocks) and induce a variety of T wave changes (including flattening, inversion, and notching).20
Figure 5. Hyperkalemia
Note the prominent, narrow, symmetric, peaked T waves in leads V3-V6.
ECG contributed by Larissa Velez, MD.
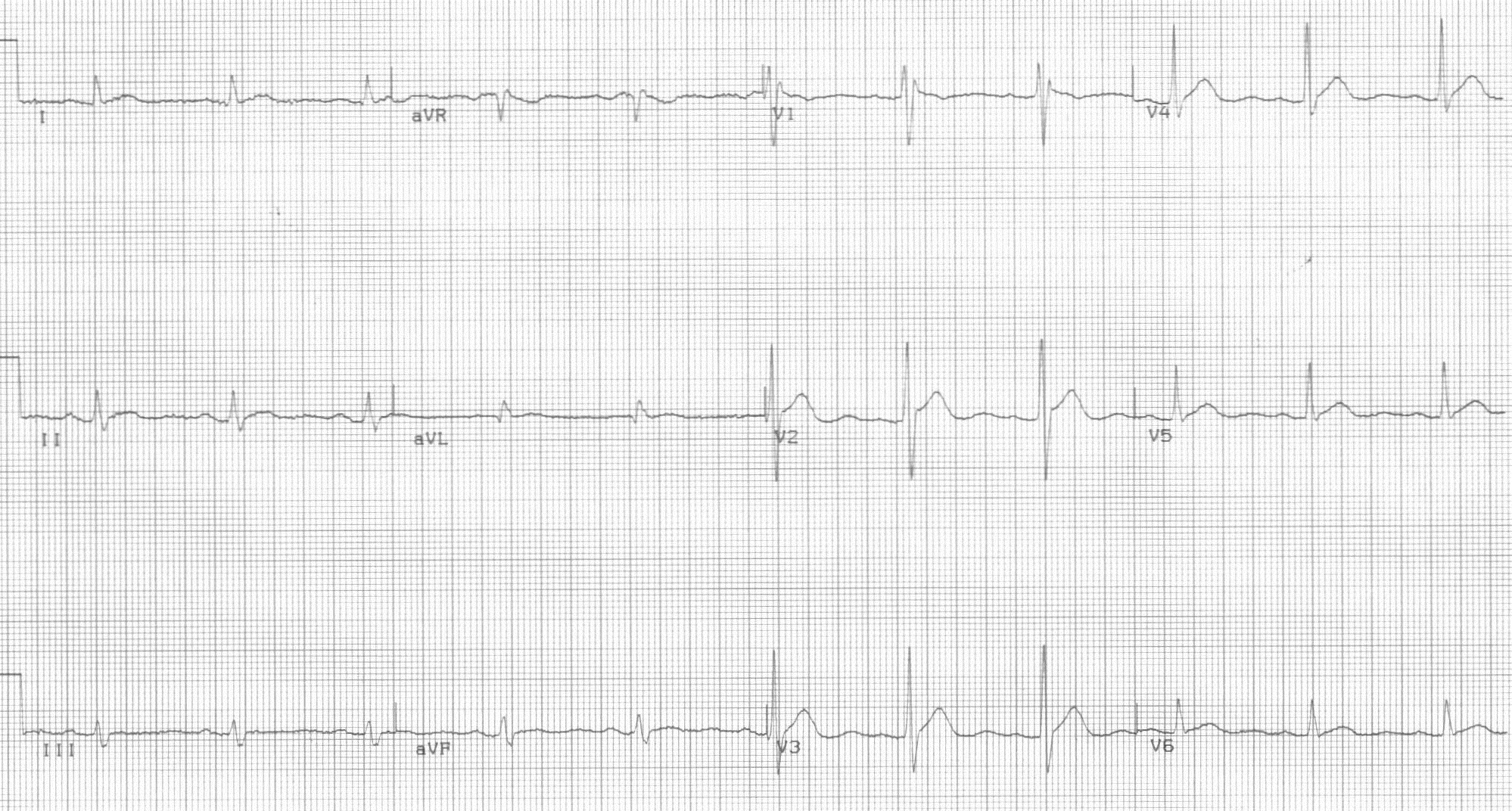
Figure courtesy of J. Stephan Stapczynski, MD.
Sodium channel blocker (SCB) toxicity may manifest as ST elevation, particularly in lead aVR. The most well studied SCBs in this context are the tricyclic antidepressants (TCAs). The classic electrocardiographic findings of TCA toxicity are sinus tachycardia, a QRS duration of more than 100 ms, and a rightward shift of the terminal 40 ms of the QRS,21 which is best demonstrated by a dominant R wave in lead aVR.22 These changes are accompanied by the anticholinergic (antimuscarinic) toxidrome and hypotension. Although cardiology literature considers a QRS duration up to 100 ms as normal, in the presence of SCB toxicity, QRS durations greater than 100 ms are associated with increased chances of seizures, and QRS durations greater than 160 ms are associated with increased chances for ventricular dysrhythmias, as described in a landmark TCA toxicity article.23
Ventricular Enlargement
Left ventricular enlargement (hypertrophy, LVH) is known to cause many ECG abnormalities and has been reported to be a cause for false-positive cardiac catheterization lab activations.7 (See Figure 7.) The electrocardiographic diagnostic criteria for LVH are poorly sensitive when compared to assessment of left ventricular mass by echocardiography.24 The most specific and most widely used criteria are the Sokolow-Lyon criteria, which confer a specificity of 100%25: the amplitude of the S in V1 + R in V5/6 > 35 mm.26
The repolarization abnormality, which can cause ST elevation in leads V1-V3 as well as T wave abnormalities (formerly known as “strain”), occurs due to the electrical remodeling (accompanied with anatomic remodeling) of the left ventricle in the setting of hypertrophy.27 The ST segment elevation seen in leads V1 to V3 in patients with LVH may mimic an anterior STEMI. It has been noted that the ST elevation seen in a STEMI is usually greater than that seen with LVH. Rather than a specific value in mm, a ratio of the ST elevation to the size of the dominant wave (either R or S) in the QRS complex in the same lead can be used to distinguish when ST elevation is within typical values for LVH. A threshold value of < 0.25 for the ST to R/S ratio has been proposed for what is normal and typical in LVH.28 However, no validated guidelines exist to address an appropriate amount of ST elevation for LVH with “strain.”29 LVH with repolarization abnormality may be indistinguishable from myocardial ischemia if no previous ECG is available for comparison.
Right ventricular (RV) enlargement is due to a high RV afterload and can be either acute or chronic (cor pulmonale). In adults, chronic RV enlargement is most commonly due to long-standing pulmonary hypertension (e.g., idiopathic pulmonary arterial hypertension, etc.) Similar to left ventricular enlargement, no sensitive criteria exist for the diagnosis of RVH. The simplest and most specific criteria is an R wave in V1 > 7 mm (or R/S in V1 > 1) and right axis deviation.25 Acute RV enlargement (“right heart strain”) does not have a large R wave in V1 but may have right axis deviation. RVH by itself rarely causes ST elevation except in some cases of acute pulmonary embolism. The ST elevation seen in acute pulmonary embolism is typically seen in leads V1 and aVR.30 A meta-analysis of ECG features in acute pulmonary embolism found that six ECG findings had significant predictive value for circulatory collapse: a heart rate > 100 beats/min, an S1Q3T3 pattern, a complete RBBB, inverted T waves in V1-V4, ST elevation in aVR, and atrial fibrillation.31 (See Figure 8.) The mechanism behind this is unclear. It has been reported that RVH can cause ST depression in V1-V3 and can mimic a posterior STEMI.32
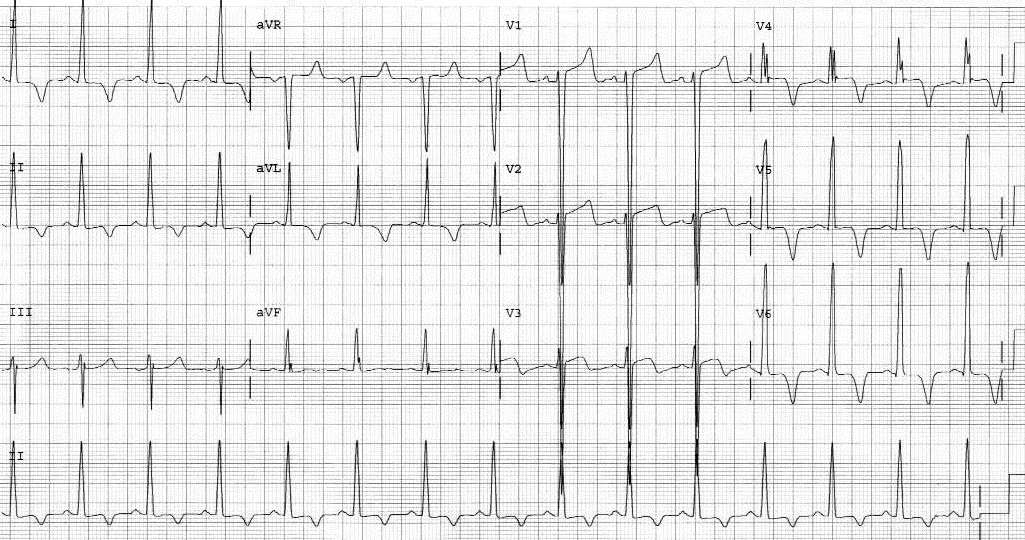
The depth of the S wave in V1 plus the height of the R wave in V5/6 is > 35 mm with ST segment and T wave changes indicative of “strain.”
Figure courtesy of J. Stephan Stapczynski, MD.
Note the sinus tachycardia (heart rate 111 beats/min), incomplete RBBB pattern, and ST elevation in lead aVR.
Figure courtesy of J. Stephan Stapczynski, MD.
Aneurysm
When a transmural infarct is not aborted by therapeutic intervention and the AMI completes itself, the myocardium is replaced by a thin, fibrous layer, which is “dyskinetic” on imaging; this is called LV aneurysm (also known as “persistent ST elevation after previous MI”). On the ECG, a left ventricular aneurysm may manifest as persistent ST elevation in the territory of a prior infarct, commonly concomitant with Q waves. (See Figure 9.) Most of these aneurysms occur at the left ventricular apex. Smith and colleagues have derived33 and validated34 a rule for differentiation of anterior LV aneurysm from acute anterior STEMI. The rule states that when the differential diagnosis is acute LAD occlusion vs. anterior LV aneurysm, if any of leads V1-V4 has a T wave amplitude to QRS amplitude ratio of > 0.36, then STEMI is likely. In general, aneurysm is favored by prominent Q waves in leads V1-V4 with corresponding diminished T wave amplitudes.
Persistent ST elevation due to LV aneurysm that developed after an anterior infarction indicated by Q waves in V2 to V4
Figure courtesy of J. Stephan Stapczynski, MD.
Takotsubo cardiomyopathy, also termed “the broken heart syndrome” or stress cardiomyopathy (SCM), refers to an apical LV aneurysm in the absence of an identifiable coronary artery culprit lesion or prior scar. It is called “Takotsubo” because it causes apical ballooning of the ventricle, in which the base of the heart contracts well, but the apex balloons out in systole. Thus it has the appearance of a Japanese octopus trap, called a “takotsubo.” First described in 1990, Takotsubo is mainly a disease of older women (mean age 66 years and 89.8% women35). It has been reported to be the cause of troponin positive STEMI in 1-2% of that population. The ECG of Takotsubo is identical to an anterior STEMI (most commonly), which may also present with diffuse, bizarre T wave inversions and a long QT. Both conditions may have increased troponins.26 In fact, 80% of patients with Takotsubo cardiomyopathy will have positive troponins; therefore, these patients should still undergo angiography to evaluate for an obstructive lesion.35
Thailand (Brugada) Syndrome
As mentioned previously, Brugada syndrome is considered a part of the ERS spectrum36 but is a distinct clinical entity with potentially lethal consequences. For many years, a “sleeping sickness” was described in Southeast Asia (known as “Lai Lai” in Thailand), in which young men would scream in the middle of the night and suddenly die. In 1992, Brugada syndrome was initially described by the Brugada brothers as an RBBB with persistent ST elevation in patients presenting with syncope and dysrhythmic events.37
It should be noted, however, that Brugada syndrome is defined as the ECG pattern in addition to symptoms (syncope, palpitations, sudden death). If seen in isolation, the Brugada ECG pattern is referred to as Brugada sign (whose significance is yet indeterminate). It is thought that the Brugada syndrome is the cause of death in about 4-5% of sudden death cases, particularly in young males. After genotyping, the most common mutation observed was in SCN5A,25 a sodium channel. Although this channel’s predominant contribution is during phase 0 and 1 of the fast myocyte action potential,38 it is postulated that this mutation leads to an unopposed outward potassium current.
The ECG pattern manifests an indistinct R wave in which the ST segment has a gradual downslope, such that at 40 ms after the ST takeoff, the increase in amplitude is < 4 mm.39 This abnormal ST segment may be mistaken for ST elevation, particularly in patients who present with ventricular dysrhythmias. There are two (previously considered to be three) ECG patterns in Brugada syndrome. (See Figure 10.) The most recognized Brugada ECG phenotype, Type 1, has ST elevation in V1-3 of a “coved” variety25 and is frequently identified as a right bundle branch block. Type 2 has “saddle back” ST morphology with > 2 mm ST elevation. The Brugada ECG phenotype may be transient40 and exacerbated by the presence of sodium channel blockers, by fever, or have no identifiable precipitant. Patients with Brugada syndrome must be evaluated for internal cardioverter defibrillator (ICD) placement to prevent sudden death.39 Since the absolute risk of death in patients with isolated Brugada sign is unknown, emergency providers should recommend follow-up with cardiology.
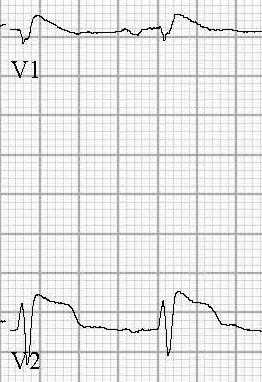
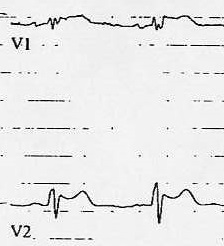
Left figure: Type I Brugada pattern with coved ST segment.
Right image: Type II Brugada pattern with saddle back ST segment
Figures courtesy of J. Stephan Stapczynski, MD.
Inflammation (Myopericarditis)
The classic ECG changes associated with acute pericarditis, a relatively rare clinical entity, are diffuse ST elevation and PR depression. (See Figure 11.) When evaluating for these two findings, it is crucial to remember that the isoelectric line in the ECG is the TP segment. The morphology of ST elevation of pericarditis can mimic that of an infarct and can cause both confusion and consternation to the emergency physician.
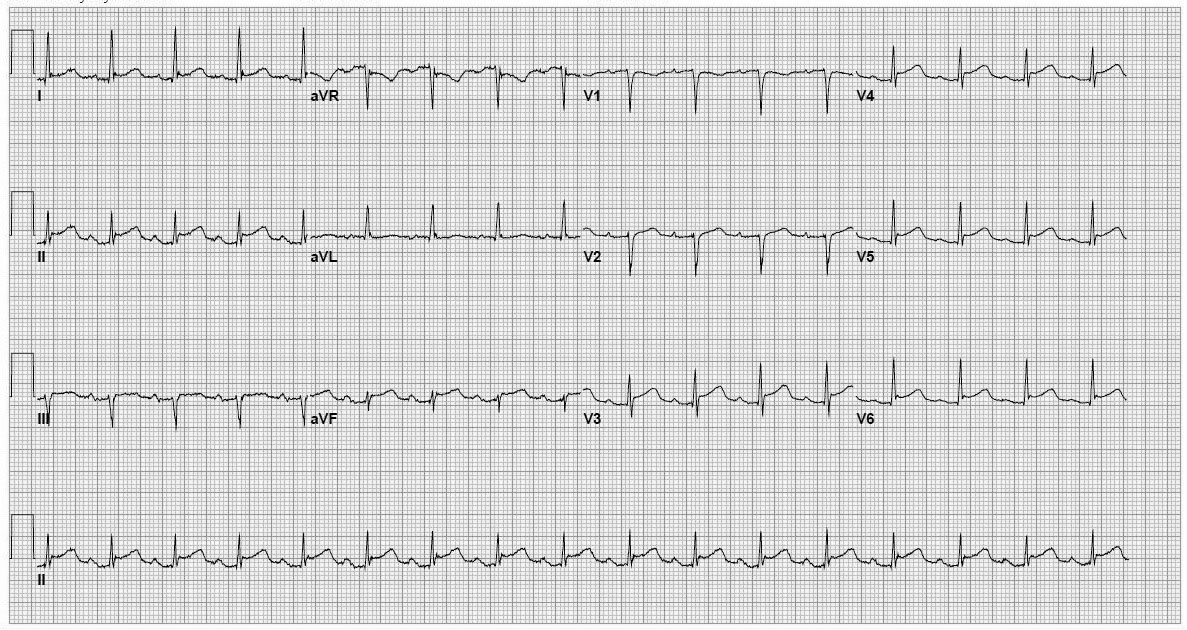
Diffuse ST elevation most prominent in leads I1, II, and V4-V6, without reciprocal changes (ST segment depression in leads opposite those with ST segment elevation), Spodick’s sign (downsloping TP segment in lead II), and PR segment depression (best seen in lead II).
ECG courtesy of Walter Green, MD, FACEP.
The diagnosis of AMI is favored if any of the following two ECG findings are present41: any ST depression (other than V1 or aVR); or ST elevation in lead III that is greater than the amplitude of ST elevation in lead II.42 In pericarditis, ST segment elevation is seen most commonly and most prominently in the inferior and high lateral leads. Notably, if there is inferior ST elevation, the existence of ST depression in aVL favors inferior wall MI over pericarditis.43 In other words, any suggestion of reciprocal changes should raise the clinician’s suspicion for STEMI.
In general, PR depression is only reliably seen in viral acute pericarditis, is transient, and must be seen in multiple leads.44 The “checkmark” or “RT sign” seen in some cases of pericarditis describes the appearance of a checkmark-like deflection at the terminal portion of the QRS joining the T wave. Spodick’s sign, a downsloping of the TP segment and best seen in lead II and the lateral precordial leads, is seen in about 80% of pericarditis patients.45 In addition, the ECG findings of STEMI are often dynamic compared to those of pericarditis, which are unlikely to change in the ED. Prolongation of the QRS complex and shortening of the QT interval also can be seen in patients with STEMI and not pericarditis.46 One final way to differentiate between pericarditis and early repolarization, calculate the ratio of the height of the onset of the ST segment/T wave amplitude in V6. If this ratio is > 0.25, acute pericarditis is likely.47
Osborn (J) Waves
In 1953, Osborn performed an experimental hypothermia observation study on dogs, cooling them to 23°C. Osborn noted a pattern on the ECG that conferred a bad prognostic sign and a likelihood of progression to VF arrest.48 Initially it was postulated that the Osborn wave was not necessarily a current of injury similar to that of an infarct, but, rather, due to an impeded elimination of CO2. Osborn waves, synonymous with “J waves,” are brief positive deflections at the junction of the QRS complex and ST segment. Osborn waves are most commonly observed in hypothermia, and may be seen in hypercalcemia, vasospastic myocardial ischemia, and brain injury. (See Figure 12.) In the setting of hypothermia, beyond the obvious low body temperature, the J wave is usually seen in the context of bradycardia. The cellular basis for this J wave was clarified as a difference in the action potential propagation between epicardial M cells and endocardial cells49 and is thought to be the same mechanism behind ERS. As mentioned previously, the clinical implication of the J wave may be more far-reaching than previously considered. Once thought to be “simply” an RBBB pattern, the J wave associated with Brugada syndrome may carry a risk of sudden death. Differentiating a J wave from ST elevation seen in an infarct has to do with the continued downward deflection seen after a J wave but not seen in ST elevation.
Figure 12. Osborn Waves
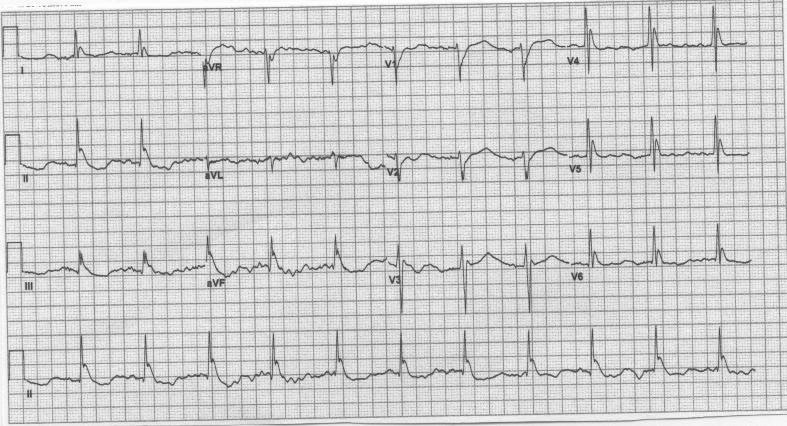
Non-Thrombotic Vasospasm
ST elevation due to vasospasm has an identical morphology to that of a true STEMI; however, it is not due to coronary artery endoluminal plaque rupture and intraluminal thrombosis. Vasospastic angina (once termed “Prinzmetal,” or variant angina) occurs due to coronary artery vasospasm (hence giving ST elevation in a given coronary territory). Soon thereafter, or after the administration of nitrates, the chest pain and ST elevation resolve. Infarcts due to plaque rupture typically do not have resolution of ST elevation after the administration of nitrates or calcium channel blockers. In the ED, this often is difficult to sort out, so patients with chest pain and ST elevation in a vascular territory always should have cardiology involved early for the decision to perform catheterization.
ST elevation due to cocaine occurs due to a similar mechanism. Vasospasm occurs due to the adrenergic effects, which also increase myocardial oxygen demand. Chest pain is a common complaint of patients who present to the ED after using cocaine. The COCHPA study group determined that there was no clinical parameter to predict which patients were at very low risk for cocaine-induced MI.50 As cocaine-associated MI is a well-established entity, and cocaine users are at an increased risk for accelerated atherosclerosis,51 all patients with cocaine use and chest pain should be treated as potentially having acute coronary syndrome.52
CONCLUSION
Emergency physicians evaluate patients with chest pain every day. In patients with ECG changes consistent with a STEMI, the primary goal is to salvage myocardium by rapid reperfusion by an interventional cardiologist (when available). However, in many clinical scenarios, non-ischemic causes of ST segment elevation need to be considered. A clinical context and a thorough evaluation of the ECG will often provide clues to some of these STEMI mimics.
REFERENCES
- Brady WJ, Perron AD, Martin ML, et al. Cause of ST segment abnormality in ED chest pain patients. Am J Emerg Med 2001;19:25-28.
- O’Gara PT, Kushner FG, Ascheim DD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of ST-elevation myocardioal infarction: A report of the Americal College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013;127:e362-425.
- Li RA, Leppo M, Miki T, et al. Molecular basis of electrocardiographic ST-segment elevation. Circ Res 2000;87:837-839.
- Samson WE, Scher AM. Mechanism of S-T segment alteration during acute myocardial injury. Circ Res 1960;8:780-787.
- Lilly L. Pathophysiology of heart disease: A collaborative project of medical students and faculty. Philadelphia: Lippincott Williams & Wilkins; 2011:1605477230.
- Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm 2015;12:1852-1866.
- Larson DM, Menssen KM, Sharkey SW, et al. “False-positive” cardiac catheterization laboratory activation among patients with suspected ST-segment elevation myocardial infarction. JAMA 2007;298:2754-2760.
- Macfarlane PW, Antzelevitch C, Haissaguerre M, et al. The early repolarization pattern: A consensus paper. J Am Coll Cardiol 2015;66:470-477.
- Deshpande A, Birnbaum Y. ST-segment elevation: Distinguishing ST elevation myocardial infarction from ST elevation secondary to nonischemic etiologies. World J Cardiol 2014;6:10.
- Smith SW, Khalil A, Henry TD, et al. Electrocardiographic differentiation of early repolarization from subtle anterior ST-segment elevation myocardial infarction. Ann Emerg Med 2012;60:45-56.
- Smith S. Formula to differentiate normal variant ST elevation (early repolarization) from anterior STEMI. Dr. Smith’s ECG Blog. June 16, 2013. Available at: http://hqmeded-ecg.blogspot.com/2013/06/here-is-link-to-full-text-of-article-in.html. Accessed April 5, 2016.
- Antzelevitch C, Yan GX, Viskin S. Rationale for the use of the terms J-wave syndromes and early repolarization. J Am Coll Cardiol 2015;57:1587-1590.
- Neeland IJ, Kontos MC, de Lemos JA. Evolving considerations in the management of patients with left bundle branch block and suspected myocardial infarction. J Am Coll Cardiol 2012;60:96-105.
- Sgarbossa EB, Pinski SL, Barbagelata A, et al. Electrocardiographic diagnosis of evolving acute myocardial infarction in the presence of left bundle branch block. N Engl J Med 1996;334:481-487.
- Mattu A. ECG Case of the Week. Available at: https://www.youtube.com/watch?v=jGQajcVgYPM. July 23, 2012. Accessed May 9, 2016.
- Smith SW, Dodd KW, Henry TD, et al. Diagnosis of ST-elevation myocardial infarction in the presence of left bundle branch block with the ST-elevation to S-wave ratio in a modified Sgarbossa rule. Ann Emerg Med 2012;60:766-776.
- Tabas JA, Rodriguez RM, Seligman HK, et al. Electrocardiographic criteria for detecting acute myocardial infarction in patients with left bundle branch block: A meta-analysis. Ann Emerg Med 2008;52:329.
- Meyers HP, Limkakeng AT Jr, Jaffa EJ, et al. Validation of the modified Sgarbossa criteria for acute coronary occlusion in the setting of left bundle branch block: A retrospective case-control study. Am Heart J 2015;170:1255-1264.
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol 2008;3:324-330.
- Nishi SP, Barbagelata NA, Atar S, et al. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol 2006;39:298-300.
- Wolfe TR, Caravati EM, Rollins DE. Terminal 40-ms frontal plane QRS axis as a marker for tricyclic antidepressant overdose. Ann Emerg Med 1989;18:348-351.
- Harrigan RA, Brady WJ. ECG abnormalities in tricyclic antidepressant ingestion. Am J Emerg Med 1999;17:387-393.
- Boehnert MT, Lovejoy FH Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med 1985;313:474-479.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990;81:1144-1146.
- Fuster V, Walsh RA, Harrington RA. Hurst’s The Heart. New York: McGraw-Hill Education; 2012.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949;37:161-186.
- Schocken D. Electrocardiographic left ventricular strain pattern: Everything old is new again. J Electrocardiol 2014;47:595-598.
- Armstrong EJ, Kulkarni AR, Bhave PD, et al. Electrocardiographic criteria for ST-elevation myocardial infarction in patients with left ventricular hypertrophy. Am J Cardiol 2012;110:977-983.
- Birnbaum Y, Alam M. LVH and the diagnosis of STEMI — how should we apply the current guidelines? J Electrocardiol 2014;47:655-660.
- Shy BD, Gutierrez A, Strayer RJ. Bedside ultrasound to evaluate pulmonary embolism masquerading as ST elevation myocardial infarction. J Emerg Med 2015;49:703-704.
- Shopp JD, Stewart LK, Emmett TW, et al. Findings from 12-lead electrocardiography that predict circulatory shock from pulmonary embolism: Systematic review and meta-analysis. Acad Emerg Med 2015;22:1127-1137.
- Smith S. A 50-something male with dyspnea. Dr. Smith’s ECG Blog. 3/2/2015. Available at: http://hqmeded-ecg.blogspot.com/2015/03/a-50-something-male-with-dyspnea.html. Accessed May 10, 2016.
- Smith SW. T/QRS ratio best distinguishes ventricular aneurysm from anterior myocardial infarction. Am J Emerg Med 2005;23:279-287.
- Klein LR, Shroff GR, Beeman W, et al. Electrocardiographic criteria to differentiate acute anterior ST-elevation myocardial infarction from left ventricular aneurysm. Am J Emerg Med 2015;33:786-790.
- Templin C, Ghadri JR, Diekmaan J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med 2015;373:929-938.
- Ali A, Butt N, Sheikh AS. Early repolarization syndrome: A cause of sudden cardiac death. World J Cardiol 2015;7:46-475.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol 1992;20:1391-1396.
- Brunton LL, Chabner BA, Knollmann BC, eds. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill Education/Medical; 2011.
- Bayes de Luna A, Brugada J, Baranchuk A, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: A consensus report. J Electrocardiol 2012;45:433-442.
- Hoogendijk MG, Opthof T, Postema PG, et al. The Brugada ECG pattern: A marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrythm Electrophysiol 2010;3:283-290.
- Farzad A. ST-elevation: When it’s NOT a STEMI. Dallas, TX. Jan. 2, 2016.
- Mattu A. Amal Mattu’s ECG Case of the Week — July 20, 2015. ECG Weekly. July 20, 2015. Available at: https://ecgweekly.com/2015/07/amal-mattus-ecg-case-of-the-week-july-20-2015/. Accessed Feb. 28, 2016.
- Bischof JE, Worrall C, Thompson P, et al. ST depression in lead aVL differentiates inferior ST-elevation myocardial infarction from pericarditis. Am J Emerg Med 2016;34:149-154.
- Mattu A. ECG Workshop Part II. University of Maryland Emergency Medicine. Available at: https://umem.org/files/uploads/1505111254_ECG_workshop_part_II.pdf.
- Chaubey VK, Chhabra L. Spodick’s sign: A helpful electrocardiographic clue to the diagnosis of acute pericarditis. Perm J 2014;18:e122.
- Rossello X, Wiegerinck RF, Alguersuari J, et al. New electrocardiographic criteria to differentiate acute pericarditis and myocardial infarction. Am J Med 2014;127:233-239.
- Ginzton LE, Laks MM. The differential diagnosis of acute pericarditis from the normal variant: New electrocardiographic criteria. Circulation 1982;65:1004-1009.
- Osborn JJ. Experimental hypothermia: Respiratory and blood pH changes in relation to cardiac function. Am J Physiol 1953;175:389-398.
- Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation 1996;93:372-379.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine-associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med 1994;1:330-339.
- Patrizi R, Pasceri V, Sciahbasi A, et al. Evidence of cocaine-related coronary atherosclerosis in young patients with myocardial infarction. J Am Coll Cardiol 2006;47:2120-2122.
- McCord J, Jneid H, Hollander JE; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Management of cocaine-associated chest pain and myocardial infarction: A scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation 2008;117:1897-1907.
Several conditions can mimic an acute infarction by producing ST segment elevation. It is important for the emergency physician to recognize these conditions to appropriately manage patients in the ED and to better distinguish acute infarction from other conditions on the ECG.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.