Pediatric Syncope: Current Status of Diagnostic Evaluation and Management
AUTHORS
Thomas Dalton, MD, Clinical Assistant Professor of Emergency Medicine, Stanford Department of Emergency Medicine, Stanford, CA
N. Ewen Wang, MD, Professor of Emergency Medicine, Associate Director, Pediatric Emergency Medicine, Stanford School of Medicine, Stanford, CA
PEER REVIEWER
Christopher J. Haines, DO, FAAP, FACEP, Chief Medical Officer, Children’s Specialized Hospital, New Brunswick, NJ; Associate Professor of Pediatrics and Emergency Medicine, Drexel University College of Medicine, Attending Physician, St. Christopher’s Hospital for Children, Philadelphia, PA
Children may present to the emergency department with a potential syncopal event. Although the presentation is unusual, everyone fears missing a cardiac issue. The authors present a concise review, focusing on the history, physical exam, and ECG, of how to evaluate and manage a child with syncope, differentiating other mimics and discussing the current therapeutic approach to the most common diagnosis.
— Ann M. Dietrich, MD, FAAP, FACEP
Definition of the Problem
Syncope is defined as a rapid-onset, transient loss of consciousness, with complete spontaneous recovery to the patient’s baseline mental status due to transient cerebral hypoperfusion.1 Syncope and pre-syncope in the pediatric patient are common presenting problems for the emergency provider (EP). Most presentations are benign, and only approximately 2% of patients will have a serious underlying cardiac cause of the event.2,3 The challenge for the EP is to identify these rare life-threatening presentations, differentiating syncope from other potential serious disorders, all while balancing appropriate testing to avoid unnecessary interventions. This article will address the epidemiology of syncope in pediatric patients, the causes and classification of syncope, as well as disorders that mimic it, and provide an algorithm for the evaluation, management, and disposition of pediatric patients who present with syncope to the emergency department (ED). It will not address other causes of transient loss of consciousness, such as head injuries, cerebrovascular accidents, or toxic ingestions.
Epidemiology
Syncope is a common presenting problem in the pediatric population, representing slightly less than 1% of all pediatric ED presentations. Approximately 40% of girls and 20% of boys experience a syncopal event by the age of 18 years.2-5 The incidence increases with age and peaks in adolescence, with girls presenting more often than boys.2,3,6 The underlying pathophysiology processes leading to syncope are similar in both adult and pediatric patients, but the incidence of more serious cardiac arrhythmias and structural abnormalities is higher in adult patients.7 Of undifferentiated pediatric patients presenting to the ED with complaints of syncope or pre-syncope, the majority will have neurally mediated (predominantly vasovagal) and orthostatic syncope, approximately 9% will have neurological disorders including seizures, and approximately 2% will have an underlying cardiac cause.2-3 Of patients with an underlying cardiac cause, excluding patients with a known preexisting cardiac disorder, less than 1% of total patients will have a newly diagnosed cardiac cause as the etiology of the event.2 The remaining patients typically have a mixture of diagnoses, including psychogenic events, intoxications, and metabolic disorders.3
Clinical Presentations of Syncope
Syncope is classically divided into three distinct types — neurally mediated, orthostatic, and cardiovascular. (See Table 1.) The diagnosis is dependent on a thorough history and exam, including an understanding of the symptoms related to the event. However, children may not be able to describe the symptoms thoroughly, and the event may occur so suddenly that the patient and observers may not be able to recall when postural tone was lost.
Table 1. Classification of Syncope1,72 |
|
Neurally Mediated Syncope |
|
Vasovagal:
|
|
Situational:
|
|
Syncope Caused by Orthostatic Hypotension |
|
Primary autonomic failure:
|
|
Secondary autonomic failure:
|
|
Drug-induced orthostatic hypotension:
|
|
Hypovolemia:
|
|
Cardiac Syncope |
|
Arrhythmias:
|
|
Structural disease:
|
In all cases, the underlying pathophysiology is due to a transient decrease in cerebral blood flow secondary to decreased blood pressure. This in turn is due to a decrease in cardiac output and/or peripheral vascular resistance.
Neurally Mediated Syncope. Neurally mediated syncope, otherwise known as neurocardiogenic or reflex syncope, refers to a heterogeneous group of disorders caused by inappropriate cardiovascular reflexes that lead to vasodilation, bradycardia, and/or decreased contractility in response to a specific trigger. This includes both vasovagal and situational syncope.
Vasovagal syncope is the most well-known cause in this group and is the most common cause of syncope in the pediatric population.3 It is more common in females than males and tends to be more prevalent in adolescence and less prevalent in patients younger than 10 years of age.3 Diagnostically, a provider should be cautious in ascribing the cause of a syncopal event to vasovagal syncope in a younger child, as more serious processes, such as a cardiac cause of syncope or a syncope mimic (seizure or breath-holding spell), are more common in this age group. Vasovagal syncope typically results from a specific trigger that increases parasympathetic tone and decreases sympathetic tone. Common triggers include emotional stress, pain, trauma, being in a crowded or warm environment, or phobias such as to blood or needles. Often, these triggers are potentiated by hunger, exhaustion, intercurrent illness, and orthostatic stress, such as from hypovolemia, prolonged standing, and/or positional changes. Characteristically, there is a prodrome of symptoms lasting seconds to minutes secondary to activation of the autonomic nervous system. Common symptoms include gastrointestinal upset or abdominal pain, nausea, warmth, general weakness, lightheadedness, or tunnel vision.
In the case of situational syncope, the trigger is a decrease in preload that causes an exaggerated parasympathetic response due to increased stimulation of heart mechanoreceptors. Examples include cough/sneeze syncope, syncope with heavy weight lifting secondary to the Valsalva maneuver, micturition syncope, and post-exercise syncope. In all cases, the loss of consciousness typically lasts from 5-20 seconds but should not last for longer than several minutes. During the event the patient may appear pale, ill, and diaphoretic. Immediately following the syncopal event, a patient’s mental status should be normal but there is typically a recovery phase of 5-30 minutes during which the patient will feel weak, fatigued, and dizzy. Complaints of general weakness, nausea, and headaches are common.8 The event can recur if the patient sits or stands suddenly from a supine position.
The diagnosis of neurally mediated syncope is a clinical one, made after excluding other life-threatening causes. Treatment is supportive. Increased salt and water intake is recommended. Education on appropriate actions to take when triggers are encountered is also important to abort episodes and includes lying down when the prodromal symptoms first occur and performing isometric contractions of the extremities, such as folding the arms and crossing the legs.
Orthostatic Syncope. With a positional change from sitting or supine to upright, blood volume is redistributed to the legs and splanchnic circulation. This causes a decrease in preload with a resultant decrease in cardiac output. The decrease in cardiac output provokes a cascade of compensatory reflexes that lead to an increase in sympathetic tone and decrease in parasympathetic tone. This results in an increase in vascular resistance, venous return to the heart, and cardiac contractility, all of which help normalize the blood pressure. In certain conditions in which the body’s compensatory reflexes are impaired or in cases of moderate to severe hypovolemia, there can be a large drop in blood pressure, resulting in orthostatic pre-syncope or syncope. Clinically, the onset of symptoms is not immediate and is accompanied by lightheadedness, dizziness, weakness, and blurring of the vision. Orthostatic vital signs traditionally have been used to confirm a diagnosis of orthostatic syncope and determine the presence of hypovolemia. Unfortunately, there is variation in the definition of vital sign abnormalities used to define what constitutes “positive” orthostatic vital signs, i.e., orthostatic hypotension with regard to the vital signs used, the timing of measurements, and which positions to measure them.9 The most widely endorsed definition of orthostatic hypotension is a decrease in systolic blood pressure of at least 20 mmHg or diastolic blood pressure of 10 mmHg measured within three minutes when moving from a supine to standing position.10 This does not account for measurements of the heart rate seen in other definitions, such as postural increase in heart rate being regarded as a positive indication for orthostatic hypotension or if the patient is symptomatic when changing position.9 Most literature on this subject has focused on adult patients, and overall orthostatic vital sign changes have been found to be poorly sensitive and specific in detecting hypovolemia.11-13
In children, data on orthostatic vital signs are sparse. One study that compared clinically dehydrated children in the ED to euvolemic controls found an increase in heart rate of > 20 beats per minute (bpm) to be 81% sensitive and specific for dehydration based on clinical signs, but found no clinically significant change between groups with regard to orthostatic blood pressure measurements.14 Another study of euvolemic asymptomatic pediatric patients found that there was a wide range of postural vital sign changes with a significant overlap with the traditional definition of positive orthostatic vital signs.15 In children, an increase in the heart rate > 20 bpm when moving from supine to standing is reasonably sensitive to detect clinical hypovolemia, but changes in blood pressure have been found to correlate poorly. Additionally, the specificity of a change in vital signs to distinguish between hypovolemic and euvolemic patients is poor. There is more clinical utility if an orthostatic position change reproduces a patient’s symptoms and if there are other clinical findings supporting a diagnosis of hypovolemia.
The major cause of orthostatic hypotension in pediatric patients is hypovolemia.6,7 In most cases, the focus should be on identifying and addressing the underlying cause of hypovolemia such as anemia or dehydration.
Except for postural orthostatic tachycardia syndrome (POTS), other causes of autonomic dysfunction and drug-induced orthostatic hypotension are rare in comparison to the adult population. Postural orthostatic tachycardia syndrome is an uncommon disorder that predominantly affects adolescents and young adults, with females being more commonly affected than males.17,18 The exact mechanisms are unclear but it is thought to be due in part to autonomic dysfunction causing orthostatic intolerance.16-18 The diagnosis of POTS is established with three criteria: 1) frequent symptoms, such as lightheadedness, general weakness, fatigue, palpitations, and malaise when moving from a recumbent to standing position; 2) a sustained increase in the heart rate ≥ 30 bpm (≥ 40 bpm for patients ages 12-19 years) for more than 30 seconds with a postural change from supine to standing; and 3) lack of the 20 mmHg drop in systolic blood pressure characteristic for orthostatic hypotension.16,19 Typically, symptoms should be present for at least three months.19 At baseline, the standing heart rate can be > 120 bpm, and other symptoms, such as headaches, abdominal pain or bloating, urinary symptoms, nausea, fatigue, and sleep disturbances, are common even without positional changes.16,18,20 This chronic condition typically resolves in a patient’s mid-20s, but it can be frustrating for patients and caregivers, as it significantly adversely affects the patient’s quality of life.16,18,20
The differential diagnosis is broad and includes thyrotoxicosis, cardiac arrhythmias, hypoadrenalism, anemia, hypovolemia, or medication side effects, such as from beta-blockers or diuretics. This is a difficult diagnosis to make in the ED and is considered a diagnosis of exclusion. To rule out common conditions that have similar features, it is reasonable to obtain orthostatic vital signs, an electrocardiogram, complete blood count, electrolytes, and thyroid function studies if there is clinical concern. If the diagnosis is suspected but uncertain, then tilt-table testing to confirm orthostatic intolerance can be obtained on an outpatient basis by the patient’s regular provider; it is not indicated emergently. Further testing with cardiac event monitoring, an echocardiogram, and more sophisticated laboratory testing, such as testing for serum norepinephrine levels, can be deferred to the outpatient setting on an individualized basis.16
Treatment is supportive, and long-term management is best individualized and done by a multidisciplinary team. For an acute decompensation resulting in a presentation to the ED, rehydration with 1 to 2 liters of normal saline is reasonable. In the short term, even if the diagnosis is suspected but not confirmed, education, behavior modification, regular exercise, and an increase in salt and water intake are first-line treatments.16,21
Cardiac Syncope. Cardiac causes of pediatric syncope are rare but important, representing approximately 2% of all cases of pediatric syncope that present to the ED.2,3 Fortunately, sudden cardiac death is rare in the pediatric population, occurring in approximately 1 to 10 in 100,000 children per year in the western world, and 61% will have had no known prior history of cardiac disease.22,23 Of those pediatric patients with sudden cardiac death, 45% will have had some antecedent symptoms and approximately 8% will have had a previous episode of syncope.23 Clinical signs and symptoms that are more suggestive of a cardiac etiology are similar to those in the adult population. The presence of palpitations, occurrence while supine, and a limited prodrome ≤ 5-10 seconds are more predictive of a cardiac etiology, as is a history of underlying cardiac disease and sudden onset without classic neurally mediated prodromal symptoms.3,24,25 Syncope that occurs with exertion is more likely to be cardiac in nature compared to syncope that occurs just after exertion.25,26 This is an important distinction to make, as it is not uncommon to experience lightheadedness and occasionally faint after very vigorous exertion, especially if deconditioned. In contrast, syncope that occurs during exertional activity is more worrisome except in situations in which there is a clear Valsalva maneuver, such as in heavy weight lifting. Worrisome exam findings include the presence of a pathological murmur such as a holosystolic murmur, a diastolic murmur, a systolic crescendo-decrescendo murmur, or a harsh or grade 3 or higher murmur.27,28
Syncope from cardiac causes generally can be grouped into obstructive and arrhythmogenic causes. Obstructive causes result in mechanical impediment to cardiac output. This can include both intrinsic cardiac problems, such as structural abnormalities from unrepaired or repaired congenital heart disease, aortic stenosis, or hypertrophic cardiomyopathy (HCM), and extrinsic cardiac causes, such as pulmonary hypertension or a large pulmonary embolism. Arrhythmogenic causes include both brady and tachyarrhythmias, such as sick sinus syndrome, high-degree atrioventricular (AV) blocks such as Mobitz Type II second-degree and third-degree AV blocks, supraventricular tachycardia (SVT), ventricular tachycardia (VT), and ventricular fibrillation (VF). There can be an overlap of both arrhythmogenic and obstructive causes in patients with underlying heart disease, such as patients with hypertrophic cardiomyopathy being at risk for syncope both from aborted episodes of VT and VF as well as from outflow obstruction. This also holds true for patients with some forms of repaired structural congenital heart disease. The EP also should be aware of several congenital cardiomyopathies and channelopathies that can cause syncope. These include congenital long QT syndrome, Brugada syndrome, HCM, and arrhythmogenic right ventricular dysplasia (ARVD).
Patients with a prolonged QT interval can present with episodes of syncope that represent self-terminated episodes of polymorphic ventricular tachycardia. QT prolongation occurs both congenitally, such as in long QT syndrome, or can be secondary to a variety of causes, such as the use of QT-prolonging medications, hypothyroidism, and electrolyte abnormalities including hypokalemia, hypomagnesemia, or hypocalcemia. Congenital long QT syndrome occurs when there are mutations in cardiac ion channels that predispose a patient to polymorphic ventricular tachycardia and sudden cardiac death.29 This disorder can be inherited or arise from de novo mutations and can present with variable penetrance. The most common inherited form is Romano-Ward syndrome, which presents in an autosomal dominant fashion with purely arrhythmic manifestations. More rare forms include Timothy syndrome, which is associated with autism, developmental delay, structural cardiac defects, and syndactyly; Andersen–Tawil syndrome, which is associated with facial and limb defects; and Jervell and Lange-Nielsen syndrome, in which there is an association with sensorineural hearing loss. As the QT interval varies with the heart rate, it should be corrected for this using Bazett’s formula where the corrected QT interval (QTc) is equal to the QT interval divided by the square root of the R to R interval. This is done automatically on ECGs but is prone to inaccuracy and should be hand calculated in leads II or V5 and averaged over several cycles. In healthy pediatric patients, the upper normal limit of the QTc interval is 450 msec, and in patients with congenital long QT syndrome, the average QTc interval is 482 msec.30-32 More than one-third of patients referred to a specialty clinic for congenital long QT syndrome with a prior diagnosis of this disorder were found not to actually have it. This is caused by a combination of factors, including frequent incorrect calculation of the QTc interval, diagnosing the syndrome based on “borderline” elevated QTc intervals, and/or attribution of vasovagal syncope to a prolonged QTc arrythmatic event.32,33 In unexplained syncope without a clear alternative cause, such as classic vasovagal syncope, a diagnosis can be established if secondary causes of QTc prolongation are ruled out and the QTc interval is > 480 msec on repeated ECGs.33 Once secondary causes of a prolonged QTc interval are ruled out, the probability of congenital long QT syndrome being present can be determined by the long QT score (see Table 2), which factors in the QTc interval, other ECG abnormalities such as T-wave alternans in which there is variation in the morphology of the T-wave, T-wave notching, family and clinical history, and the response of the QTc interval from stress testing to derive the probability of congenital long QT syndrome. A score of ≤ 1 rules out the diagnosis, a score of 1.5-3.0 is considered intermediate probability of the diagnosis, and a score of ≥ 3.5 is considered diagnostic.33,34 In borderline or uncertain cases, consultation with a cardiologist is recommended to guide further workup and the need for admission. Initial management is with beta-blockers and implantable cardiac defibrillator placement for patients with prior cardiac arrests or recurrent episodes of syncope while on beta-blockade.
Table 2. Long QT Scoring Criteria34 |
|
|
ECG Findings |
Points |
|
QTc interval |
|
|
3 |
|
2 |
|
1 |
|
QTc interval at 4th minute of recovery from an exercise stress test ≥ 480 |
1 |
|
Torsades de pointes |
2 |
|
T wave alternans |
1 |
|
Notched T waves in 3 leads |
1 |
|
Low heart rate for age |
0.5 |
|
Clinical History |
|
|
Syncope |
|
|
2 |
|
1 |
|
Congenital deafness |
0.5 |
|
Family History |
|
|
Family member with definitive LQTS |
1 |
|
Unexplained cardiac death before age 30 years among immediate family members |
0.5 |
|
SOURCE: Author Adapted |
|
Short QT syndrome is a rare condition in which there are mutations in genes encoding for cardiac potassium channels. This can predispose the patient to episodes of VT or VF and sudden cardiac death. This is diagnosed with the presence of a QTc interval ≤ 330 or with a QTc interval ≤ 360 but with a known pathogenic mutation, family history of the syndrome, a family member with sudden death at age ≤ 40 years, or if the patient survived a VT/VF episode in the absence of heart disease.33 Treatment with an implantable cardiac defibrillator is indicated in patients with a history of resuscitated cardiac arrest, documented episodes of VT, or in patients with the syndrome with a family history of sudden cardiac death.33
Catecholaminergic polymorphic ventricular tachycardia is a rare inherited disorder in which there are mutations in genes that encode for proteins that control calcium release in the myocardium. This can be inherited in an autosomal dominant or autosomal recessive fashion and predisposes the patient to sudden cardiac death from polymorphic ventricular tachycardia.33 It is triggered by high adrenergic states, such as with exertion or emotional stress. The baseline ECG will be normal and there will be no structural abnormalities on cardiac imaging. The diagnosis likely will be established on cardiac monitoring after a patient presents with a concerning history for an arrhythmogenic cause of syncope. A classic presentation would be a child who has repeated episodes of fainting during times of emotional stress or fear. Treatment is with avoidance of strenuous exertion, competitive sports, stressful activities, and the use of beta-blockers, and placement of an implantable cardiac defibrillator.
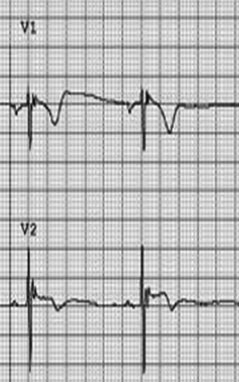
Brugada syndrome occurs secondary to mutations in cardiac sodium channels that predispose the patient to ventricular fibrillation and sudden cardiac death. Structurally, the heart will be normal. This is inherited in an autosomal dominant fashion and tends to have a higher incidence in males and patients of Asian descent.33 Syncope in these patients is a concerning sign and is a risk factor for sudden cardiac death.34 ECG findings can be variably present in an individual with this disorder, but the pathognomonic finding includes the presence of a “coved” or “shark-fin” ST segment elevation > 2 mm in > 1 of V1-V2 followed by T-wave inversions.33,35 (See Figure 1.) A variety of factors — including fevers, alcohol, drug use, and certain medications such class 1 antiarrhythmics (flecainide, procainamide, etc.), certain anesthetics (bupivacaine, propofol, etc.), and some psychotropics (lithium, nortriptyline) — can predispose patients with this disorder to arrhythmias. Placement of an implantable cardiac defibrillator is indicated in patients with Brugada syndrome if they have survived a resuscitated cardiac arrest, have documented episodes of VT, or have had episodes of syncope that are thought to be secondary to ventricular arrhythmias.33
Figure 1. Brugada Syndrome |
|
Note the “coved” or “shark-fin” ST elevation in V1, ST elevation in V2, and T-wave inversions in both leads |
 |
|
Courtesy of Dr. Victor Froelicher. |
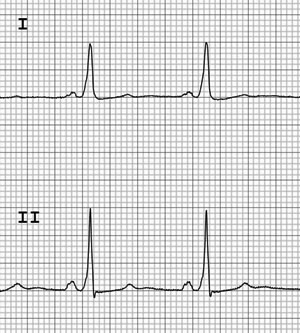
Wolff-Parkinson-White (WPW) syndrome occurs when there is the presence of an accessory pathway between the atria and ventricles that causes an AV reentrant tachycardia (AVRT); this can result in palpitations and syncope, and in rare cases can degenerate into VF in the presence of atrial fibrillation and cause sudden cardiac death.36,37 In pediatric patients, the incidence is approximately 0.23 cases per 1,000 patient-years.38 Key ECG features include a slurring of the initial portion of the QRS complex known as a delta-wave, reflecting early ventricular depolarization from conduction down the accessory pathway. This will cause a short PR interval < 120 ms and QRS prolongation > 110 ms. (See Figure 2.) The presence of ST segments and T-wave changes is normal and reflects that abnormal ventricular depolarization. Recurrent tachyarrhythmias are managed long term with antiarrhythmics, nodal blockade agents, and catheter ablation of the accessory pathway.37
Figure 2. Wolff-Parkinson-White Syndrome |
|
Note the up-sloping delta-wave at the beginning of the QRS complex, wide QRS complex, and short PR interval. |
 |
|
Courtesy of Dr. Victor Froelicher. |
Hypertrophic cardiomyopathy results from genetic mutations in cardiac proteins resulting in disordered cardiac muscle growth and asymmetric ventricular hypertrophy. It has an incidence in children of 0.3-0.5 cases per 100,000 individuals per year and is inherited in an autosomal dominant pattern with variable penetrance, but rarely can arise from de novo mutations.39,40 Patients with this disorder can be predisposed to arrhythmias, such as AV blocks, atrial fibrillation, and ventricular tachycardia, and can develop syncope, ischemia, and heart failure. It also is the leading cause of sudden cardiac death in young athletes in the United States.41
Syncope in patients with this disorder can occur from common etiologies, such as vasovagal syncope, but can also occur from a variety of other causes, such as cardiac arrhythmias and dynamic outflow obstruction.39,40 At baseline, there is asymmetric ventricular hypertrophy, which narrows the aortic outflow tract and can limit cardiac output. Dynamic outflow obstruction occurs during certain conditions when the cardiac output is transiently decreased because of the narrowing of this tract. This can occur with a decrease in afterload, a decrease in the ventricular chamber size, or an increase in the rate of blood flow through the outflow tract.39 With an increase in blood flow through the outflow tract, the mitral valve leaflets will be pulled into and block the tract in a phenomenon called systolic anterior motion, and hence will decrease cardiac output. When dynamic outflow obstruction occurs, it can lead to syncope and cardiac ischemia but also can result in mitral regurgitation and signs of heart failure.
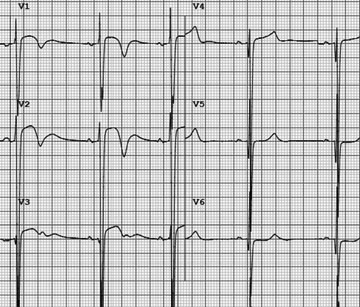
To prevent these complications, it is important to avoid and treat conditions that decrease the preload, such as dehydration, and treat conditions that increase the heart rate or contractility, such as atrial fibrillation with a rapid ventricular response. In this disorder, the ECG will be abnormal in 97% of cases.42 Key ECG features include excessive voltage, especially in the precordial leads, left atrial enlargement, and the presence of one or both of deep “dagger-like” Q waves and T-wave inversions. (See Figure 3.) Patients may have a harsh crescendo-decrescendo murmur heard loudest at the left upper sternal border that reflects outflow tract obstruction, and in cases of mitral regurgitation, a holosystolic murmur can be heard as well.40 In contrast to most other murmurs, the crescendo-decrescendo seen in hypertrophic cardiomyopathy will increase with maneuvers that decrease the preload and thereby cause more dynamic outflow obstruction. This can be seen as the murmur increases in severity when moving from sitting to standing or with a Valsalva maneuver. Treatment of this disorder can include beta or calcium blockade, septal reduction, and implantation of an automatic cardiac defibrillator. In unclear cases or when the etiology is more suggestive of a cardiac cause, such as with associated chest pain, palpitations, or little prodrome, consultation with a cardiologist is recommended to guide further evaluation and typically will include admission for cardiac monitoring, rest and stress echocardiography, and Holter monitoring after discharge.40
Figure 3. Hypertrophic Cardiomyopathy |
|
Note the excessive precordial voltage, deep T-wave inversions, and lateral deep Q waves |
 |
|
Courtesy of Dr. Victor Froelicher. |
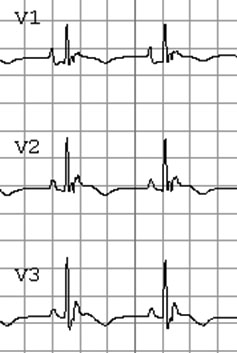
Arrhythmogenic right ventricular dysplasia, also known as arrhythmogenic right ventricular cardiomyopathy, is a genetic disorder in which mutations in cardiac proteins lead to fibrofatty replacement of the right ventricular myocardium with subsequent right ventricular dysfunction.43 This can arise from de novo mutations but can be inherited, typically in an autosomal dominant fashion with variable penetrance. The disordered myocardium predisposes the patient to arrhythmias, such as ventricular tachycardia and ventricular fibrillation, which can result in syncope and sudden death. This disorder accounts for approximately 4% of sudden cardiac deaths in young athletes in the United States.41 It typically presents in adolescence and young adulthood with combinations of palpitations, syncope, chest pain, and/or shortness of breath, but sudden cardiac death can be the first manifestation.43 The diagnosis is complex and is based on family history, ECG abnormalities, tissue biopsies, and the presence of right ventricular dysfunction on either echocardiography, MRI, or angiography.44 The ECG in children with this disorder will be abnormal in most cases. These abnormalities can include the presence of precordial T wave inversions in leads V1-3 in patients > 14 years of age, borderline QRS widening > 110 msec in V1-3, small deflections following the QRS complex called epsilon waves, complete or incomplete right bundle branch blocks, signs of right ventricular failure such as dominant R-waves in the right precordial leads, frequent premature ventricular contractions, and episodes of ventricular tachycardia with a left bundle branch block morphology.44,45 (See Figure 4.) Treatment is variable and includes restriction from vigorous exercise, antiarrhythmics, cardiac ablation, and implantation of a cardiac defibrillator.46
Figure 4. Arrhythmogenic Right Ventricular Dysplasia |
|
Note the R > S wave in V1-3 indicating right ventricular enlargement, epsilon-waves following the QRS complex, right bundle branch block morphology, and T-wave inversions. |
 |
|
Courtesy of Dr. Anne M. Dubin. |
One special population that warrants discussion is patients with underlying heart disease, whether congenital, such as tetralogy of Fallot or hypoplastic left heart syndrome, or acquired, such as due to myocarditis. It is out of the scope of this article to discuss congenital heart disease in depth; however, generally, because of ventricular dysfunction, structural abnormalities, and scarring, these patients are at a higher risk of sudden cardiac death compared to the general population, with sudden cardiac death accounting for 15-25% of deaths in patients with structural congenital heart disease.22,23 Predominately these deaths are due to arrhythmias.47 This is a heterogeneous group of disorders, but unexplained syncope can be worrisome for an arrhythmia or other complication. From an ED perspective, when a pediatric patient who has a known history of cardiac disease presents with syncope, consultation with the child’s cardiologist is recommended to guide the decision for further management and disposition.
Syncope Mimics
Several disorders closely mimic syncope, including seizures, psychogenic events, toxic/metabolic disorders, and breath-holding spells. (See Table 3.) In many of these cases, the loss of consciousness will be prolonged and/or the patient will have not recovered to baseline spontaneously within minutes, excluding the patient from the definition of true syncope.
Table 3. Mimics of Syncope1,3 |
|
|
SOURCE: Author adapted |
Seizures. Approximately 3% of pediatric patients initially diagnosed with syncope will be found to actually have a seizure disorder.6 Differentiating the two can be difficult, as seizure-like activity occurs in 16-34% of patients who present with syncope, and between 7-12.9% of patients with prior diagnosis of seizures will be found to have syncope.6,26,48,49 Several features can help differentiate a seizure from syncope. (See Table 4.) In comparison to a seizure, the shaking/jerking activity that is seen with syncope occurs after the loss of postural tone and not before, and the duration of the seizure-like activity is also shorter, lasting only 5-15 seconds, and typically is not rhythmic.50 Tongue lacerations also are seen more often in seizure than syncope (33% sensitive, 96% specific).51 Other specific findings differentiating seizures from syncope include head turning with the event (43% sensitive, 97% specific), unusual posturing (35% sensitive, 97% specific), and urinary incontinence (17% sensitive, 74% specific).52,53 Two key features that are very sensitive for seizures are the presence of a postictal state in which there is prolonged confusion for more than several minutes in contrast to syncope (94% sensitive, 69% specific) and if there are any abnormal behaviors noted by bystanders (92% sensitive, 67% specific).52 Features that are more common in syncope but not seizures include the presence of prodromal pre-syncopal symptoms such as lightheadedness, blurry vision, or weakness; the occurrence with a clear trigger, such as a blood draw or prolonged sitting or standing; palpitations or chest pain before the event; and if the patient can recall losing consciousness.52 At times the provider will be uncertain as to the diagnosis, so a parallel workup for both etiologies can be performed. Even if there is uncertainty, most patients can be discharged home safely from the ED with appropriate referral if a cardiac etiology of syncope or an acutely life-threatening cause of a seizure are ruled out.
Table 4. Clinical Features Differentiating Syncope from Seizures50-53 |
||
|
Clinical Feature |
Syncope |
Seizures |
|
Provocation stimulus |
Frequent |
Rare |
|
Duration of loss of consciousness |
Seconds to 1-2 minutes |
Greater than 1-2 minutes |
|
Involuntary movements |
Last < 15 seconds, not synchronized |
Last minutes, rhythmic, synchronized |
|
Timing of movements |
After loss of tone |
Before loss of tone |
|
Tongue biting |
Rare |
Common |
|
Incontinence |
Rare |
Common |
|
Postictal confusion and recovery period |
Not present, recovery occurs in seconds to a min |
Present, recovery takes many minutes to hours |
|
SOURCE: Author adapted |
||
Breath-holding Spells. Breath-holding spells are a common cause of loss of consciousness in children and occur in 4.6% of pediatric patients.54 They normally occur between the ages of 6 to 18 months. Twelve percent can occur before 6 months of age, and they are rare beyond 4-7 years of age.55-57 Breath-holding spells are thought to be due to an interplay of exaggerated centrally mediated reflexes leading to increased vagal nerve stimulation, inappropriate pulmonary reflexes, and a possible component of autonomic dysregulation.56,57 Typically, they are triggered by painful stimuli, a frightening event, or frustration in the child. Clinically, this can present with either a cyanotic or pallid breath-holding spell or even as a mixed disorder with features of both. In the cyanotic form, after the trigger, the child cries, has a forced expiration, turns blue, and then loses consciousness. Stiffening of the body followed by limpness can follow this.56,57 A pallid breath-holding spell occurs when there is a painful stimulus to the child. This can be minor such as to a ground-level fall or light blow to the body or head. The child turns pale and diaphoretic with little to no crying and then loses consciousness and goes limp. The onset can be delayed for up to 30 seconds after the initial traumatic event. In both cases, the child typically recovers within 1-2 minutes but may be tired and sleep for the following hour. In a small percentage, there may be jerking and incontinence.55,57 In contrast to a seizure, these jerking/stiffening movements occur at the terminal portion of the event, not the beginning, and color changes occur before and not after any changes in tone and posture.57 There is no known increased risk of developing seizures or associated cerebral injury with these spells.55 The diagnosis is clinical; however, a pallid breath-holding spell or a complex breath-holding spell with jerking motions may be impossible to distinguish from arrhythmogenic syncope or a seizure, respectively. Treatment of cyanotic breath-holding spells is supportive with reassurance and education of the caregivers, as these events can recur and are anxiety provoking.
Psychiatric Causes of Syncope. Syncope can occur in the pediatric patient from hyperventilation, such as from a panic attack, or from a conversion disorder. Syncope from a conversion disorder, otherwise known at pseudosyncope, occurs when there is an apparent loss of consciousness without an actual loss of consciousness. It is thought to be due in part to a physical manifestation of psychological distress.58 Estimates of the prevalence vary.58,59 Pseudosyncope usually occurs in adolescents and younger adults, can occur with emotional stress, typically only occurs with an audience, and a significant percent have comorbid psychiatric conditions.58-61 Patients often have atypical presentations that do not fit an underlying pathophysiological cause of syncope or a seizure, such as prolonged periods of apparent loss of consciousness/unresponsiveness lasting more than the seconds to minutes expected with seizures or syncope. Episodes can occur multiple times a day, and the patient can have an unusually calm demeanor or “la belle indifference” in the face of their apparent symptoms.58 Also in contrast to syncope, the majority of patients with pseudosyncope will have their eyes closed during the event vs. having their eyes open (7% vs. 97%).61 Some overlap exists between patients with pseudosyncope and actual syncope, with 28% of patients in one series exhibiting both syncope and pseudosyncope.58 In cases in which the diagnosis is in question, referral for further testing is warranted. Although not in the scope of this article, in general, treatment is supportive and includes educating the patient and caregiver that these spells are involuntary, which helps prevent the patient from feeling stigmatized that he/she is “faking” or purposefully causing the symptoms. Referral to a psychiatrist or psychotherapist is beneficial, but care should be taken to frame it as a referral to a specialist who can help identify and deal with stress and other triggers of these events, as patients and caregivers are reluctant to accept a psychiatric explanation of their symptoms and focus more on the physical rather than psychological aspects.
History and Examination
The goal of ED evaluation is to identify life-threatening causes of syncope, exclude mimics, and risk stratify the patient for occult cardiac pathology when the diagnosis is unclear. The cornerstone of this evaluation will be the history, as most pediatric patients who present with syncope will appear well on arrival and have a normal physical examination.3 This will be supported by a focused exam and judicious diagnostic testing.1,3,47,62
Details should be obtained from both the patient and anyone who witnessed what occurred before, during, and after the event. This should include where the event occurred, the nature of the activity when the event occurred, if the patient was standing or seated, if there were any positional changes, and the patient’s emotional state, hydration status, and last meal.
It is important to ask about prodromal symptoms, including the presence of classic symptoms associated with neurally mediated syncope (such as gastric symptoms, dizziness, weakness, tunnel vision) and more concerning symptoms (such as palpitations, chest pain, or trouble breathing, which are worrisome for a cardiac etiology).24,25,47 Information should be obtained from witnesses on how long the event lasted, if there was any seizure-like behavior or post-event confusion, and if so, for how long. The EP should ask about past syncopal events as well as the context for these events. Prior evaluations and treatments and any history of cardiac or neurological problems should be assessed, and the EP should inquire about the child’s exertional status and if there have been any recent changes. A complete medication history should be taken, especially for birth control pills, cardiac medications such as nodal blockade agents, blood pressure medications, diuretics, and medications that can prolong the QT interval.
A family history is critically important and should include a history of any members with cardiac or neurological problems such as hypertrophic obstructive cardiomyopathy, arrhythmias, seizures, or a history of congenital deafness. The presence of family members who have died suddenly when young for unexpected reasons is worrisome and should prompt further workup.26 Examples include a history of sudden infant death syndrome or a family member who was a strong swimmer but drowned.
The physical exam should focus on identifying any cardiac abnormalities such as a pathological murmur. Findings such as café au lait spots, dysmorphic features, hearing loss, developmental delay, and abnormalities found on the neurological exam can be indicative of either an underlying neurological disorder predisposing to a seizure or a congenital abnormality predisposing to a cardiac arrhythmia. The patient’s hydration status should be assessed, as well as signs such as scleral pallor or inappropriate tachycardia that may represent underlying anemia. (See Table 5.)
Table 5. Findings Concerning for a Cardiac Cause of Syncope2,7,24-26,47 |
|
|
SOURCE: Author adapted |
Diagnostic Studies
Numerous studies have found a low diagnostic yield to obtaining screening tests routinely in pediatric patients who present with syncope unless supported by a strong clinical history and exam.3,26,63,64 The only routinely recommended tests are an ECG and, for all age-appropriate females, a pregnancy test. Unless supported by the history and exam, other tests are expensive, invasive, and may cause false positives that can lead to further unnecessary testing and caregiver anxiety. These include routine laboratory studies such as blood glucose, hemoglobin, electrolytes, and renal function; imaging studies such as CT scans of the head; chest X-rays; and specific cardiac and neurological tests such as echocardiograms, outpatient cardiac monitors, tilt-table testing, or EEGs.3,26,63,64 Often, EPs may be concerned that serious pathology will be missed unless a more extensive evaluation is performed. This is unfounded, and there are algorithms that limit unnecessary testing other than ECG and pregnancy testing in pediatric patients who present to the ED with syncope, except if suggested by the history and exam, without increasing morbidity or mortality.65,66
The prevalence of ECG abnormalities diagnostic of the cause of syncope varies based on the patient population studied. In undifferentiated pediatric patients presenting to the ED with syncope, a causative abnormality based on the ECG is found in < 1% of patients vs. approximately 15% in patients referred to a cardiology clinic.26,63 Still, it is recommended that an ECG be obtained on all patients who present to the ED with syncope, as it is noninvasive, relatively inexpensive, and helps screen for occult cardiac causes of syncope.1,26,65,66 Concordance between EPs and pediatric cardiologists in interpreting pediatric ECGs obtained for all causes is variable, with differing interpretations in 13-40% of cases.67,68 Fortunately, most differences in interpretation are not clinically significant. Data looking at the accuracy of emergency providers to correctly interpret ECGs specifically obtained in cases of pediatric syncope are limited, but small studies have found that EPs tend to overcall abnormalities, specifically in regard to possible QTc prolongation, prompting the authors to recommend consultation with a pediatric cardiologist prior to admitting a patient solely on the basis of an abnormal ECG.64,69
Routine screening lab work, including electrolytes, renal function, complete blood counts, and blood glucose, are of low utility unless supported by the history and physical in the otherwise asymptomatic patient with a normal exam. A loss of consciousness due to hypoglycemia will not result in spontaneous resolution of a patient’s symptoms unless corrected, although it is reasonable to obtain a blood glucose level if there is a concern that the patient is still symptomatic or if the provider is present when the event occurs. Routine screening for electrolyte abnormalities rarely will be of benefit in the otherwise asymptomatic patient without a suggestive history but should be considered when ECG abnormalities, such as a prolonged QTc interval, are present, as low levels of potassium, magnesium, and calcium can exacerbate this. Obtaining electrolytes may be indicated if there is a history of diuretic use or if the history is suggestive of gross electrolyte abnormality, such as in the patient with severe dehydration, anorexia, or bulimia. Checking hemoglobin can be useful if there is a concern for anemia, such as with an adolescent female who has heavy menses, fatigue, and scleral pallor.
Pregnancy testing should be performed on female patients of menstrual age.60,70 If the patient is pregnant, there is a different spectrum of possible life-threatening pathology to consider, including the possibilities of an ectopic pregnancy, pregnancy-related cardiomyopathy, or a pulmonary embolism. Routine testing is recommended, as patients sometimes will not be forthcoming about sexual activity because of familial, social, or cultural pressures, or may not know they are pregnant.
Cardiac event monitoring is low yield in pediatric syncope but can be obtained when there are multiple recurrent episodes of unexplained syncope or if there is a strong concern for an underlying arrhythmia.1,7 There are two main forms of cardiac event monitoring: Holter monitoring and cardiac event recorders. Holter monitors record continuous ECG data for a 24- to 48-hour period and contain a patient-activated function to mark symptomatic episodes so they can be correlated with the underlying cardiac rhythm. They have less utility when trying to capture events that occur infrequently. In these cases, a 30-day event recorder is more useful. A standard event recorder records a continuous loop of ECG data stored to memory, either when a pre-programmed arrhythmia is found or when the patient activates the recorder. In the case of patient activation, a several minute period of ECG data before and after the activation event is recorded. Thirty-day event recorders have more diagnostic utility than a Holter monitor in diagnosing underlying arrhythmias, but the downsides include complexity of use, limited memory, which can be filled by artifact if the pre-programmed arrhythmia alarm triggers incorrectly, reducing the diagnostic yield.
Echocardiogram is recommended when there is a strong clinical concern for an underlying structural cardiac abnormality, such as if the exam is suggestive of a pathological murmur or the ECG is concerning for ischemia or an underlying structural disorder such hypertrophic cardiomyopathy or arrhythmogenic right ventricular dysplasia.1,7
Tilt-table testing seeks to induce neurally mediated reflex syncope in a controlled setting and can do so with sensitivities of approximately 61-69% and specificities of 92-94%.1 It is not indicated to confirm a diagnosis of classic neurally mediated syncope, in which the history, exam, and an ECG alone are sufficient to establish a diagnosis. It can be obtained in patients with recurrent episodes of syncope of unclear etiology when a cardiac cause has been reasonably excluded to help confirm a diagnosis of neurally mediated syncope as the etiology. Other indications include to differentiate neurally mediated syncope from orthostatic syncope or confirm orthostatic intolerance such as in POTS, and to differentiate syncope from seizures or pseudosyncope.1 Tilt-table testing is not indicated emergently and can be ordered at the discretion of outpatient providers at the point of follow-up.
Neuroimaging and EEGs should not be obtained routinely in evaluating undifferentiated syncope. Even if there is a concern for a possible seizure, emergent neuroimaging is not indicated unless there is a concern for a prolonged altered level of consciousness or there is a concern for a space-occupying lesion such as in a patient with focal neurological deficits. Non-emergent neuroimaging with an MRI and EEG testing can be done safely on an outpatient basis by a neurologist.71
Differential Diagnosis and Management
The diagnostic evaluation for any pediatric patient presenting with syncope should proceed in a stepwise manner. (See Figure 5.) The first step should be to stabilize the patient and address any obvious life-threatening causes. Next, the EP should perform a detailed history and exam, obtaining an ECG on all patients and giving a pregnancy test to all females of menstrual age. If pregnant, then evaluation for an ectopic pregnancy, pulmonary embolism, or cardiomyopathy should be considered. Orthostatics can be obtained as well and can help support the diagnosis of hypovolemia if there is a > 20 bpm increase in heart rate or the positional change reproduces the patient’s symptoms, especially if there are other supportive signs of hypovolemia or the history is suggestive of an orthostatic cause. The ECG should be reviewed for possible arrhythmogenic causes including the presence of a prolonged QTc interval, WPW, HCM, Brugada syndrome, ARVD, ischemia, and high-degree AV blocks. The possibility of a seizure should be considered. Clues for seizures include the presence of tonic-clonic movements for more than several seconds, prolonged confusion, tongue biting, and loss of bowel or bladder function. If suspicious, then appropriate neurological evaluation should be undertaken. Any other non-cardiac causes of syncope, such as intoxication with drugs or alcohol, metabolic disturbances, severe anemia or dehydration, a pulmonary embolism, migraines, or breath-holding spells, should be considered based on the history and exam. Further testing and treatment as appropriate are indicated. The presence of any “red flag” signs for an occult cardiac cause should be reviewed and should prompt admission and/or cardiology consultation if present. Red flags include a concerning ECG, chest pain, trouble breathing and/or palpitations before the event, a lack of prodrome or occurrence while supine, occurrence during exertion, an abnormal cardiac exam, or a strong history of familial sudden death, familial congenital heart disease, and possible inherited cardiac abnormalities or arrhythmias.2,7,24-26,47 After excluding most mimics and other life-threatening causes, consideration should be given to the diagnosis of a neurally mediated event. This will represent the vast majority of presentations but should be considered a diagnosis of exclusion. If so, treatment is supportive. If no clear diagnosis is present after a detailed history and exam, then further focused assessment as indicated should be taken based on provider concern for other etiologies.
Figure 5. Proposed Diagnostic Algorithm for Evaluating Pediatric Syncope in the ED |
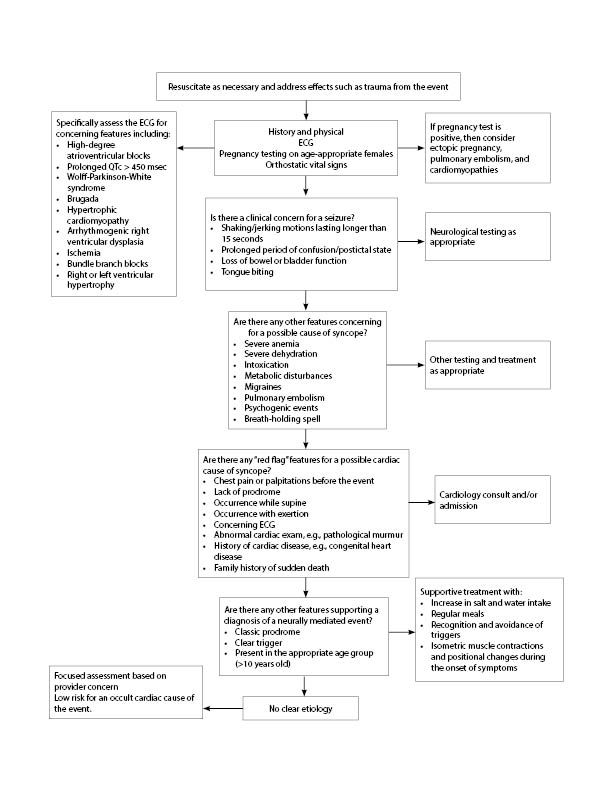 |
Disposition
An algorithmic approach will identify the vast majority of serious causes of syncope. Children with a normal examination, normal vital signs, and no red flag signs or symptoms concerning for an occult cardiac cause can be discharged safely with close follow-up with a low risk of adverse events.2,26,65,66 Patients who present with a likely obstructive cause of syncope, such as aortic stenosis, should be admitted, as should patients with clear ECG abnormalities (such as Brugada syndrome, ARVD, etc.) that increase the likelihood of life-threatening arrhythmias. In certain cases, there will be uncertainty if the syncopal event was caused by a cardiac event. In these cases, discussion with a pediatric cardiologist about further testing and disposition would be appropriate. Occasionally, children will present with non-cardiac-related complaints and will be found to have incidental abnormalities, such as a possible pathological murmur, a prolonged QTc interval, or other incidental ECG abnormalities. In these cases, consultation with a pediatric cardiologist is appropriate.
Summary
Pediatric patients who present with syncope are common. Most causes are benign, but a small percentage of patients have a more serious underlying cause of syncope. An algorithmic approach to evaluation will detect the vast majority of serious causes. A detailed history, examination, and an ECG have been shown to accurately screen for the majority of occult cardiac causes of syncope. Routine screening with lab work, toxicology screens, neurological testing, and other studies should be performed only when supported by a strong clinical concern.
REFERENCES
- Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). The Task Force for the Diagnosis and Management of Syncope of the European Society of Cardiology (ESC). Eur Heart J 2009;30:2631-2671.
- Hurst D, Hirsh D, Oster ME, et al. Syncope in the pediatric emergency department – Can we predict cardiac disease based on history alone? J Emerg Med 2015;49:1-7.
- Massin MM, Bourguignont A, Coremans C, et al. Syncope in pediatric patients presenting to an emergency department. J Pediatr 2004;145:223-228.
- Anderson JB. Czosek RJ, Cnota J, et al. Pediatric syncope: National Hospital Ambulatory Medical Care Survey results. J Emerg Med 2012;43:575-583.
- Ganzeboom KS, Colman N, Reitsma JB, et al. Prevalence and triggers of syncope in medical students. Am J Cardiol 2003;91:1006-1008, A8.
- DiMario FJ Jr, Wheeler Castillo CS. Clinical categorization of childhood syncope. J Child Neurol 2011;26:548-551.
- Strickberger SA, Benson DW, Biaggioni I, et al. AHA/ACCF Scientific Statement on the Evaluation of Syncope. Circulation 2006;113:316-327.
- Fischer JW, Cho CS. Pediatric syncope: Cases from the emergency department. Emerg Med Clin North Am 2010;28:501-516.
- Naccarato M, Leviner S, Proehl J, et al. Emergency nursing resource: Orthostatic vital signs. J Emerg Nurs 2012 S;38:447-453.
- Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res 2011;21:69-72.
- McGee S, Abernethy WB 3rd, Simel DL. The rational clinical examination. Is this patient hypovolemic? JAMA 1999;281:1022-1029.
- Levitt MA, Lopez B, Lieberman ME, et al. Evaluation of the tilt test in an adult emergency medicine population. Ann Emerg Med 1992;21:713-718.
- Koziol-McLain J, Lowenstein SR, Fuller B. Orthostatic vital signs in emergency department patients. Ann Emerg Med 1991;20:606-610.
- Fuchs SM, Jaffe DM. Evaluation of the “tilt test” in children. Ann Emerg Med 1987;16:386-390.
- Castro W, Skarin R, Roscelli JD. Orthostatic heart rate and arterial blood pressure changes in normovolemic children. Pediatr Emerg Care 1985;1:123-127.
- Sheldon RS, Grubb BP, Olshansky B, et al. 2015 Heart Rhythm Society Expert Consensus Statement on the Diagnosis and Treatment of Postural Tachycardia Syndrome, Inappropriate Sinus Tachycardia, and Vasovagal Syncope. Heart Rhythm 2015;12:e41-63.
- Benarroch EE. Postural tachycardic syndrome: A heterogeneous and multifactorial disorder. Mayo Clin Proc 2012;87:1214-1225.
- Kizilbash SJ, Ahrens SP, Bruce BK, et al. Adolescent fatigue, POTS, and recovery: A guide for clinicians. Curr Probl Pediatr Adolesc Health Care 2014;44:108-133.
- Thanavaro JL, Thanavaro KL. Postural orthostatic tachycardic syndrome: Diagnosis and treatment. Heart Lung 2011;40:554-560.
- Bhatia R, Kizilbash SJ, Ahrens SP, et al. Outcomes of adolescent-onset postural orthostatic tachycardic syndrome. J Pediatr 2016;173:149-153.
- Grubb BP. Postural tachycardia syndrome. Circulation 2008;117:2814-2817.
- Ackerman M, Atkins DL, Triedman JK. Sudden cardiac death in the young. Circulation 2016;133:1006-1026.
- Winkel BG, Risgaard B, Sadjadieh G, et al. Sudden cardiac death in children (1-18 years): Symptoms and causes of death in a nationwide setting. Eur Heart J 2014;35:868-875.
- Calkins H, Shyr Y, Frumin H, et al. The value of the clinical history in the differentiation of syncope due to ventricular tachycardia, atrioventricular block, and neurocardiogenic syncope. Am J Med 1995;98:365-373.
- Alboni P, Brignole M, Menozzi C, et al. Diagnostic value of history in patients with syncope with or without heart disease. J Am Coll Cardiol 2001;37:1921-1928.
- Ritter S, Tani LY, Etheridge SP, et al. What is the yield of screening echocardiography in pediatric syncope? Pediatrics 2000;105:E58.
- Frank JE, Jacobe KM. Evaluation and management of heart murmurs in children. Am Fam Physician 2011;84:793-800.
- Etoom Y, Ratnapalan S. Evaluation of children with heart murmurs. Clin Pediatr 2014;53:111-117.
- Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with congenital long-QT syndrome. Circulation 2008;117:2184-2191.
- Rautaharju PM, Surawicz B, Gettes LS, et al. AHA/ACCF/HRS recommendation for the standardization and interpretation of the electrocardiogram: Part IV: The ST segment, T and U waves, and the QT interval. J Am Coll Cardiol 2009;53:982-991.
- Rijnbeek PR, Witsenburg M, Schrama E, et al. New normal limits for the paediatric electrocardiogram. Eur Heart J 2001;22:702-711.
- Taggart NW, Haglund CM, Tester DJ, et al. Diagnostic miscues in congenital long-QT syndrome. Circulation 2007;115:2613-2620.
- Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm 2013;10:1932-1963.
- Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long QTc syndrome. Circulation 2011;124:2181-2184.
- Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003;108:3092-3096.
- Cain N, Irving C, Webber S, et al. Natural history of Wolff-Parkinson-White syndrome diagnosed in childhood. Am J Cardiol 2013;112:961-965.
- Page RL, Joglar JA, Caldwell MA, et al. 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients with Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2016;67:e27-e115.
- Wu MH, Chen HC, Kao FY, et al. Postnatal cumulative incidence of supraventricular tachycardia in a general pediatric population: A national birth cohort database study. Heart Rhythm 2016;13:2070-2075.
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;124:e783-831.
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on the diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733-2779.
- Maron BJ, Doerer JJ, Haas TS, et al. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980-2006. Circulation 2009;119:1085-1092.
- Thompson AJ, Cannon BC, Wackel PL, et al. Electrocardiographic abnormalities in elite high school athletes: Comparison to adolescent hypertrophic cardiomyopathy. Br J Sports Med 2016;50:105-110.
- Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009;373:1289-1300.
- Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010;121:1533-1541.
- Zhang L, Liu L, Kowey PR, et al. The electrocardiographic manifestations of arrhythmogenic right ventricular dysplasia. Curr Cardiol Rev 2014;10:237-245.
- Smith W. Guidelines for the diagnosis and management of arrhythmogenic right ventricular cardiomyopathy. Heart Lung Circ 2011;20:757-760.
- Wren C. Sudden death in children and adolescents. Heart 2002;88:426-431.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999;92:15-23.
- Josephson CB, Rahey S, Sadler RM. Neurocardiogenic syncope: Frequency and consequences of its misdiagnosis as epilepsy. Can J Neurol Sci 2007;34:221-224.
- Sheldon R. How to differentiate syncope from seizure. Cardiol Clin 2015;33:377-385.
- Brigo F, Nardone R, Bongiovanni LG. Value of tongue biting in the differential diagnosis between epileptic seizures and syncope. Seizure 2012;21:568-572.
- Sheldon R, Rose S, Ritchie D, et al. Historical criteria that distinguish syncope from seizures. J Am Coll Cardiol 2002;40:142-148.
- Brigo F, Nardone R, Ausserer H, et al. The diagnostic value of urinary incontinence in the differential diagnosis of seizures. Seizure 2013;22:85-90.
- Lombroso CT, Lerman P. Breathholding spells (cyanotic and pallid infantile syncope). Pediatrics 1967;39:563-581.
- DiMario FJ Jr. Breath-holding spells in childhood. Am J Dis Child 1992;146:125-131.
- Breningstall GN. Breath-holding spells. Pediatr Neurol 1996;14:91-97.
- DiMario FJ Jr. Prospective study of children with cyanotic and pallid breath-holding spells. Pediatrics 2001;107:265-269.
- Raj V, Rowe AA, Fleisch SB, et al. Psychogenic pseudosyncope: Diagnosis and management. Auton Neurosc 2014;184:66-72.
- Heyer GL, Albert DV, Weber A, et al. Comparison of semiologies between tilt-induced psychogenic nonsyncopal collapse and psychogenic nonepileptic seizures. Epilepsy Behav 2016;62:171-175.
- Welch-Horan TB, Shenoi RP. Syncope. In: Shaw KN, et al, eds. Fleisher & Ludwig’s Textbook of Pediatric Emergency Medicine. 7th ed. Philadelphia: Wolters Kluwer; 2016:491-497.
- Tannemaat MR, van Niekerk J, Reijntjes RH, et al. The semiology of tilt-induced psychogenic pseudosyncope. Neurology 2013;81:752-758.
- Ikiz MA, Çetin, II, Ekici F, et al. Pediatric syncope: Is detailed medical history the key point for differential diagnosis? Pediatr Emerg Care 2014;30:331-334.
- Steinberg LA, Knilans TK. Syncope in children: Diagnostic tests have a high cost and low yield. J Pediatr 2005;146:355-358.
- Goble MM, Benitez C, Baumgardner M, et al. ED management of pediatric syncope: Searching for a rationale. Am J Emerg Med 2008;26:66-70.
- Guse SE, Neuman MI, O’Brien M, et al. Implementing a guideline to improve management of syncope in the emergency department. Pediatrics 2014;134:e1413-1421.
- Raucci U, Scateni S, Tozzi AE, et al. The availability and the adherence to pediatric guidelines for the management of syncope in the emergency department. J Pediatr 2014;165:967-72.e1.
- Wathen JE, Rewers AB, Yetman AT, et al. Accuracy of ECG interpretation in the pediatric emergency department. Ann Emerg Med 2005;46:507-511.
- Horton LA, Mosee S, Brenner J. Use of the electrocardiogram in a pediatric emergency department. Arch Pediatr Adolesc Med 1994;148:184-188.
- Hue V, Noizet-Yvernaux O, Vaksmann G, et al. ED management of pediatric syncope. Am J Emerg Med 2008;26:1059-1060.
- De Lorenzo RA. In: Marx JA, et al, eds. Rosen’s Emergency Medicine Concepts and Clinical Practice. 8th ed. Philadelphia: Elesvier Saunders; 2014:135-141.
- Kimia AA, Chiang VW. Seizures. In: Shaw KN, et al, eds. Fleisher & Ludwig’s Textbook of Pediatric Emergency Medicine. 7th ed. Philadelphia: Wolters Kluwer; 2016:465-471.
- Bayram AK, Pamukcu O, Per H. Current approaches to the clinical assessment of syncope in pediatric population. Childs Nerv Syst 2016;32:427-436.
Children may present to the emergency department with a potential syncopal event. Although the presentation is unusual, everyone fears missing a cardiac issue. The authors present a concise review, focusing on the history, physical exam, and ECG, of how to evaluate and manage a child with syncope, differentiating other mimics and discussing the current therapeutic approach to the most common diagnosis.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.