Evaluation of Bradycardia in the Emergency Department
AUTHORS
Tiffany Murano, MD, FACEP, Associate Professor, Department of Emergency Medicine, Rutgers New Jersey Medical School, Newark, NJ.
Harry Stark, MD, Emergency Medicine Resident, St. John’s Riverside Hospital, Yonkers, NY.
PEER REVIEWER
Sandra Najarian, MD, Attending Physician, Department of Emergency Medicine, MetroHealth Medical Center, Cleveland, OH.
My last shift was hectic, as usual. Urgent care sent over a patient for a minor complaint because his pulse was 40 beats per minute. He was otherwise healthy, and although not at first glance an “athlete,” he was fit and worked a physically demanding job. An ECG confirmed sinus bradycardia. Several different staff — triage, techs, and nurses — repeatedly asked me to treat his asymptomatic benign bradycardia.
But bradycardia is not always benign. In the Northeast, I saw several patients in whom “asymptomatic” bradycardia was the key to Lyme disease. In others, bradycardia is a marker of very high risk among patients with acute myocardial infarction.
This paper is a great review of some basics in physiology and treatment. It will be a refresher on the identification of serious arrhythmias. There are also important reminders — digoxin, calcium channel blocker or beta-blocker overdoses, and Lyme disease. It reminds us that vital signs are indeed vital, at least until we determine they can be ignored.
— Sandra M. Schneider, MD, FACEP, Editor
Introduction
Bradycardia is defined as a heart rate less than 60 beats per minute. From the healthy athlete to the most ill patient, bradycardia is a sign commonly seen in the emergency department. Bradycardia may represent a normal or incidental finding or it may be the result of a conduction abnormality that is detected during the evaluation of a symptomatic patient. The wide variety of etiologies responsible for bradycardia makes it, on its own, a typically nonspecific sign. Both intrinsic and extrinsic factors can be responsible for this often asymptomatic finding. It is crucial for the emergency physician to be able to evaluate efficiently and treat its more malignant expressions. In this article, the normal anatomy and pathophysiology of bradydysrhythmias in adults will be reviewed, followed by the latest recommendations in evaluation and management.
Physiology
Heart rates of less than 60 beats per minute do not necessarily mean that there is an underlying pathologic state. In fact, there are several instances in which bradycardia is a normal physiologic finding. For example, there is variation in the range of resting heart rates in otherwise healthy individuals. One study demonstrated heart rates as low as 46 beats per minute in men and 51 beats per minute in women in healthy individuals without cardiovascular disease.1 It also established that heart rates decrease during sleep. Resting bradycardia in trained athletes is considered a normal finding. Initially thought to be due to a decrease in intrinsic heart rate or alteration of the autonomic balance, bradycardia in athletes now is believed to be a multifactorial mechanism. Remodeling of the sinoatrial (SA) node, genetic factors, cardiac hypertrophy, and baroreflex alteration all have been hypothesized to play a role in modifying an athlete’s resting heart rate.2
The anatomy and circulation to the conducting system is important in the understanding of the pathophysiology behind bradydysrhythmias. In the normal conduction of the heart, impulses spontaneously arise from the SA node and travel through the right atrium to the atrioventricular (AV) node and then to the bundle of His in the ventricular septum. The bundle of His bifurcates into two groups of Purkinje fibers, known as the left bundle and the right bundle, which rapidly conduct impulses that initiate ventricular contraction. The heart has a complex network of sympathetic and parasympathetic innervation that affects the automaticity of the SA and AV nodes. Parasympathetic tone decreases SA node automaticity, slows AV node conduction, and slows the heart rate; sympathetic tone has the opposite effect.
The coronary arteries supply blood to the conduction system, and occlusions may lead to conduction disturbances in those anatomic areas supplied by the affected vessel. The blood supply to the SA node is provided either via the sinus-node artery, which is a branch of the right coronary artery (RCA) in approximately 65% of the population or a branch of the circumflex coronary artery in approximately 35% of the population.3,4 The blood supply to the AV node is via the AV nodal artery — a branch of the proximal posterior descending artery, which arises from the RCA in approximately 90% of the population and from the circumflex coronary artery in 10%.3,4 The RCA also supplies blood to the proximal bundle of His, while septal branches of the left anterior descending (LAD) coronary artery supply blood to the distal bundle of His, the right bundle branch, and the anterior fascicle of the left bundle branch. Septal branches of the RCA and the LAD supply blood to the posterior fascicle of the left bundle branch.5
Etiology
The etiology of bradycardia may be classified as either intrinsic or extrinsic. (See Table 1.) With intrinsic causes (such as collagen vascular diseases, infarction, infection, infiltrative diseases, and surgical trauma), the conductive tissue is replaced by fibrous tissue, resulting in conduction abnormalities. It is important to note that while infarction and infection are common etiologies, they affect AV conduction more typically and rarely cause permanent tissue injury. Extrinsic causes of bradycardia consist of autonomic-mediated syndromes that lead to increased vagal tone, pharmacologic agents, hypothyroid disease, hypothermia, hypoxia, and electrolyte abnormalities (such as hyperkalemia, hypokalemia, and hypercalcemia).
Table 1. Causes of Bradycardia
Intrinsic Causes
Collagen Vascular Diseases
- Scleroderma
- Systemic lupus erythematosus (SLE)
- Rheumatoid arthritis
Congenital/Inherited Diseases
- Myotonic muscular dystrophy
- Congenital heart disease
- Maternal SLE
- Infarction or ischemia
Infection — Primarily Affects
the AV Conduction System
- Chagas disease
- Lyme disease
- Endocarditis
- Syphilis (tertiary)
- Viral disease (i.e., parvovirus B19, coxsackievirus)
- Toxoplasmosis
- Tuberculosis
Infiltrative Diseases
- Amyloidosis
- Sarcoidosis
- Hemachromatosis
Idiopathic
Surgical Trauma
- Cardiac transplant
- Congenital defect correction
- Valve replacement
Extrinsic Causes
Autonomic-mediated Syndromes
- Carotid sinus stimulation or hypersensitivity
- Vagal stimulation (coughing, micturition, defecation, vomiting)
- Increased intracranial pressure
Pharmacologic Agents, Toxins, and Chemicals
- Anti-arrhythmic agents
(i.e., quinidine) - Βeta-adrenergic blockers
- Calcium channel blockers
- Digoxin
- Lithium
- Organophosphates
- Pilocarpine
- Muscarinic mushrooms
Hypoxia
- Obstructive sleep apnea
- Airway occlusion
- Respiratory depression, abnormalities, or diseases
Metabolic/Endocrine
- Electrolyte abnormalities (hyper- or hypokalemia, hyper- or hypocalcemia, hypermagnesemia)
- Hypothyroid disease
- Adrenal insufficiency
Hypothermia
Adapted from: Mangrum JM, DiMarco JP. The evaluation and management of bradycardia. N Engl J Med 2000;342:703-709.
SA Node Dysfunction
Sinus bradycardia occurs when there are normally conducted impulses from the SA node below a rate of 60 per minute with 1:1 AV conduction. Electrocardiogram (ECG) findings show normal PR intervals (120-200 milliseconds) that are constant, QRS complexes that are associated with each P wave, and a rate less than 60 beats per minute. (See Figure 1.) Sinus bradycardia has physiologic, pharmacologic, and pathologic etiologies. (See Table 1.)
Sick sinus syndrome (SSS) refers to a group of disorders that lead to progressive dysfunction of the SA node. The prevalence has been estimated to be approximately one in 600 patients older than 65 years of age,6 with the mean age of approximately 74 years, and it affects men and women equally.7 Possible ECG findings include sinus bradycardia, SA blocks, sinus arrest, junctional or ventricular escape rhythms, atrial fibrillation, and atrial flutter. Bradydysrhythmias (see Table 2) are needed to make a diagnosis; however, tachyarrhythmias are present in at least 50% of patients who are diagnosed with SSS, with atrial fibrillation and atrial flutter being the most common.8 In SSS, there also may be alternating tachyarrhythmias and bradydsyrhythmias known as tachycardia-bradycardia syndrome.
SA block is the transient failure of an impulse to be conducted to the atrial myocardium. This results in a pause between the P waves (with no P wave or QRS complex) that equals the length of two or more P-P intervals. Sinus arrest is the interruption of impulse generation at the SA node. This is transient in nature and results in a long pause without P waves; however, unlike SA node block, the pause seen in sinus arrest is not related to the length of the P-P interval.
Figure 1. Sinus Bradycardia
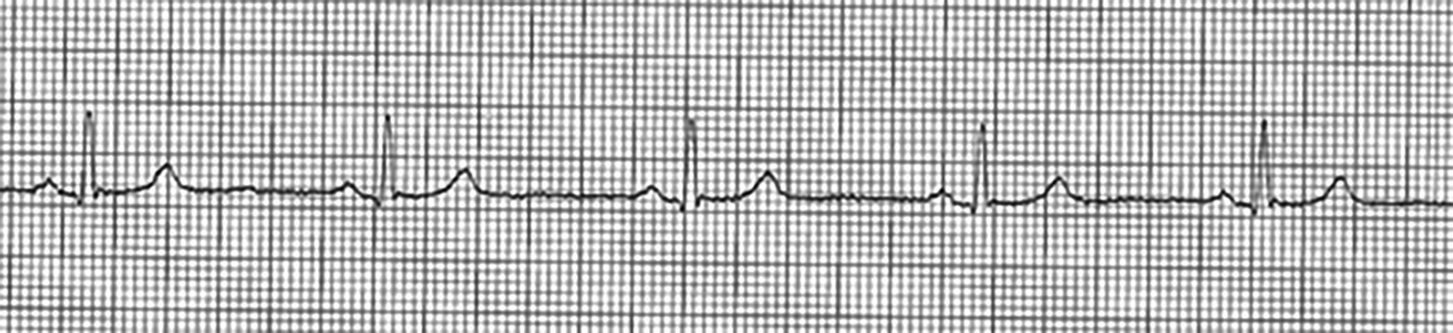
Table 2. Summary of Sinoatrial Node Disease
Condition |
Description |
|
Sinus Bradycardia |
• Normally conducted impulses from the sinoatrial node below a rate of 60 per minute with 1:1 AV conduction • Normal PR interval |
|
SA Block |
• Transient failure of an impulse to be conducted to the atrial myocardium • Pause between the P waves, with no P wave or QRS complex, that equals the length of two or more P-P intervals |
|
Sinus Arrest |
• Interruption of impulse generation at the sinoatrial node • Transient in nature and results in a long pause without P waves • Unlike SA node block, the pause seen in sinus arrest is not related to the length of the P-P interval |
|
Junctional Escape Rhythm |
• Impulse from the SA node is unusually slow or fails to generate at all • Beat generated from auxiliary pacemaker cells at the AV junction • Narrow QRS complex and a rate between 40 and 60 beats per minute |
|
Ventricular Escape Rhythm |
• Impulse from the sinoatrial node is unusually slow or fails to generate at all • Beat generated from auxiliary pacemaker cells in the ventricles • Wide QRS complex and a rate of 15 to 40 beats per minute |
AV Node Dysfunction
Disturbances in the AV node or in the bundle of His lead to delays in atrioventricular conduction. Isolated delays that are located below the bifurcation of the bundle of His typically maintain normal AV conduction and result in bundle branch or fascicular blocks. An exception to this is when there are disturbances in all three of the fascicles leading to AV conduction delays. Given its distinct connection between the atria and ventricles, focal injury to the AV node from infection, infarction, or trauma commonly causes AV conduction delays or blocks.
First-degree AV block is defined by the prolongation of the PR interval (the conduction time from the SA node to the start of ventricular depolarization) greater than 200 milliseconds (0.2 seconds). There is a QRS complex following each P wave (a 1:1 ratio) and the PR interval is constant. (See Figure 2.) First-degree AV block is encountered frequently in the clinical setting and although the most common afflicted site is the AV node, there usually are multiple points of conduction delay.9 Based on prior studies performed on young, healthy men in the military, AV block previously was thought to be a benign disorder with no clinical significance.9 However, more recent data suggest that first-degree AV block may be associated with increased risks of coronary artery disease, atrial fibrillation, heart failure, pacemaker implantation, and all-cause mortality in certain populations.9-11
Figure 2. First-Degree AV Block
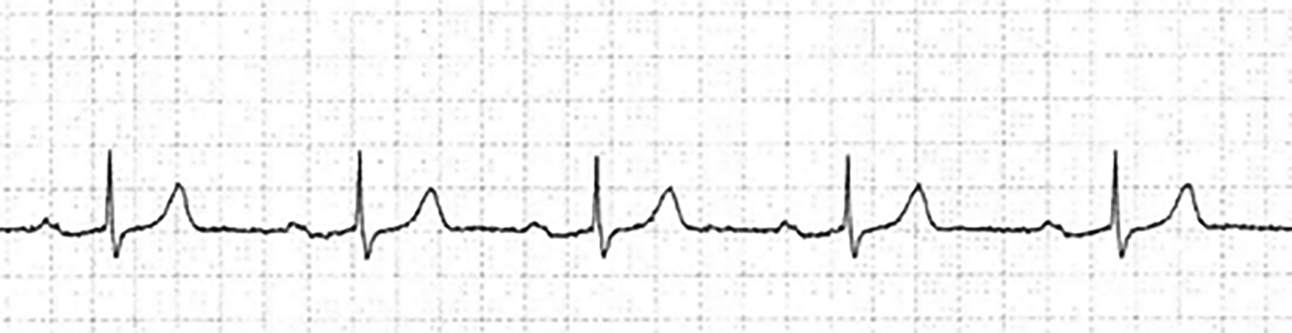
There is 1:1 AV conduction, and the PR interval is constant and > 0.2 seconds.
Second-degree AV block is the failure of consistent conduction of impulses from the atria to the ventricles in a 1:1 ratio. Second-degree AV blocks are divided into two categories: Mobitz type I (also known as a Wenckebach block) and Mobitz type II. Mobitz type I AV block is characterized by a progressive increase in the PR interval until there is a conduction failure of the atrial impulse resulting in a P wave without an accompanying QRS complex. Mobitz type I is caused most commonly by AV node delay. In Mobitz type II, the PR intervals are at regular intervals until there is an abrupt conduction failure and there is a P wave without an accompanying QRS complex. Although the PR intervals are constant, they may be normal or prolonged. Since Mobitz type II blocks most commonly are due to pathology below the AV node, the QRS complex typically is wide. However, the QRS complex may be narrow if the pathology is located in the AV node. There may be more than one blocked atrial impulse before conduction occurs. The term “high-grade” AV block refers to Mobitz type II blocks that have more than one atrial impulse that is not conducted, leading to more than one consecutive P wave before a QRS complex. When there are two consecutive P waves before a QRS complex appears, it is described as a 2:1 block; three consecutive impulses before a QRS complex is described as a 3:1 block, and so forth.
Third-degree AV block also is termed complete heart block because AV conduction is absent and atrial and ventricular activity are independent of one another. The P-P intervals remain constant, as do the R-R intervals, but there is no relationship between the two. (See Figure 3.) In this circumstance, a junctional escape pacemaker takes over and leads to ventricular contractions and, thus, determines the ventricular rate. The escape pacemaker is at a rate slower than that of the atrial pace. When the third-degree block is at the AV node, the QRS complex typically is narrow and the ventricular escape rhythm typically is between 40 to 60 beats per minute. Third-degree AV blocks that are at the infranodal level have ventricular escape rhythms slower than 40 beats per minute. When the third-degree AV block is at the bundle of His, the QRS complexes can be either narrow or wide. However, third-degree AV blocks below the bifurcation of the bundle of His produce QRS complexes that are wide.
Table 3. Summary of Atrioventricular Node Blocks
Block Type |
Description |
|
First degree |
PR interval is > 200 msec, constant 1:1 AV conduction |
|
Second degree |
|
|
Mobitz type I (Wenckebach) |
Progressive increase in PR interval until conduction failure results in P wave without a corresponding QRS complex |
|
Mobitz type II |
PR intervals are constant PR intervals may be normal or prolonged Abrupt conduction failure results in a P wave without a corresponding QRS complex May have > 1 atrial impulse not conducted, resulting in multiple P waves before QRS complex appears |
|
Third degree |
AV conduction is absent Atrial and ventricular activity are independent of one another P-P intervals are constant R-R intervals are constant |
Figure 3. Third-Degree AV Block
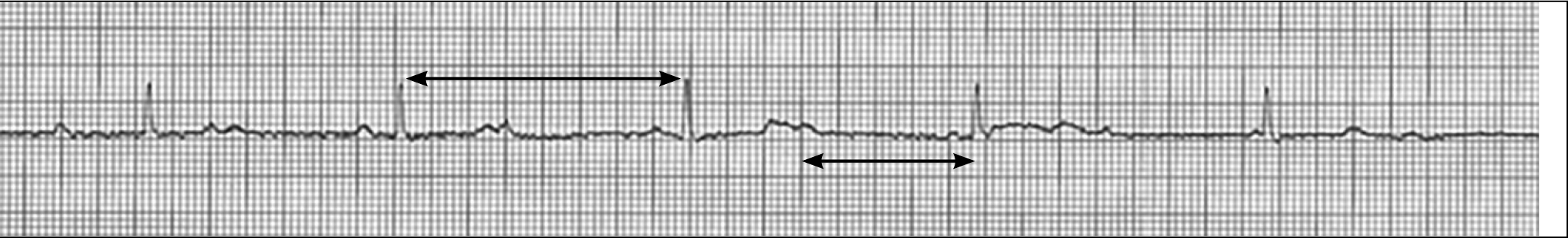
P-P intervals are constant (lower arrows). R-R intervals are constant (upper arrows). There is no relationship between the P waves and the QRS complexes.
Figure 4. Ventricular Paced Rhythm
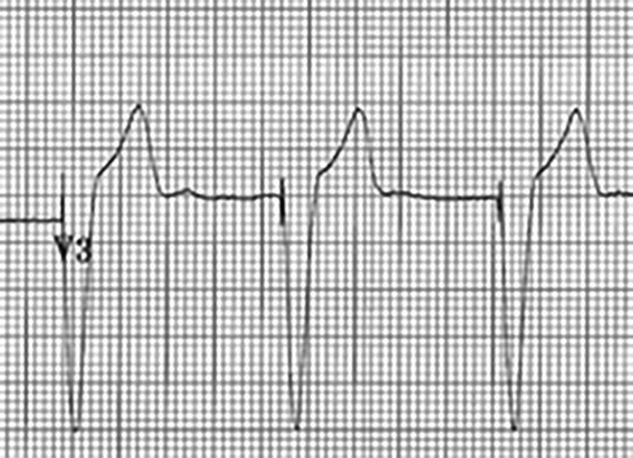
Pacer spikes are indicated before the QRS complexes.
In the setting of acute myocardial infarction (AMI), AV nodal block has significant prognostic and therapeutic significance. In AMI, first-degree AV block may occur in up to 13% of patients.12 First-degree AV block may progress to a high-degree AV block (second- or third-degree AV block) in AMI patients. The mortality rate is two to three times greater in patients who have a high-degree AV block in the AMI setting. AV nodal block in the setting of AMI frequently will present within the first 24 hours of the ischemic event and is transient the majority of the time. Third-degree AV block may occur in up to 19% of AMI patients.12 Approximately 8% of patients with inferior wall MI have third-degree AV block at the AV node.13
Junctional and Idioventricular Escape Rhythms
There are auxiliary pacemakers located in the atria, AV junction, and the ventricles. When there is a failure of the normal impulse formation or conduction, these pacemakers activate and produce escape rhythms. A junctional escape rhythm occurs if the impulse from the SA node is unusually slow or fails to generate at all; the beat is generated by the auxiliary pacemaker in the AV junction. A junctional escape beat has a narrow QRS complex and a rate between 40 and 60 beats per minute. A ventricular escape beat is generated by pacemaker cells in the ventricles and has a wide QRS complex, with a rate of 15 to 40 beats per minute.
As the name suggests, idioventricular rhythms originate from the ventricles and do not have any atrial activity. Thus, ECG findings show widened QRS complexes, typically with a rate of 30 to 50 beats per minute with no P waves. Idioventricular rhythms are present most commonly with acute ST-segment elevation myocardial infarctions. Accelerated idioventricular rhythms can be between 50 and 70 beats per minute and also are without P waves. In the setting of successful fibrinolysis of an occluded artery, an accelerated idioventricular rhythm is referred to as a “reperfusion dysrhythmia.”
Clinical Presentation
Bradycardia may be asymptomatic and, therefore, may be an incidental finding on physical examination in some patients. The heart rate typically will be less than 50 beats per minute in patients who are symptomatic. In patients whose cardiac output is decreased because of an uncompensated decrease in heart rate, symptoms such as dizziness, light-headedness, near-syncope, syncope, fatigue, and weakness may ensue. These symptoms may be constant or episodic depending on the underlying etiology. Patients who have hypoperfusion also may have signs of shock, altered mental status, chest pain/discomfort, or acute heart failure.
Evaluation
Evaluating patients who present to the ED with bradycardia begins with a focused yet thorough history in those who are asymptomatic and hemodynamically stable. Obtaining vital signs is key for the initial evaluation as well as continual monitoring. For those patients who are symptomatic or are showing signs of poor perfusion, rapid assessment of the airway, breathing, and circulation is essential. Assessment of symptom onset, frequency, and severity, as well as association with activity, is helpful in determining the etiology. Eliciting the patient’s past medical history as well as a review of the medication list, including any new medications and proper use of prescribed medications, is essential. Inquiring about potential intentional overdose of medications also is important. Ask patients with end-stage renal disease on hemodialysis about their frequency of dialysis sessions, last dialysis session, and whether the entire session was completed. A review of systems, including rash, travel history, intolerance to cold or heat, and changes in weight, may allow the emergency physician to gain valuable insight into possible underlying and treatable causes.
Physical examination findings will help guide management. Assess for signs of hypoperfusion or hemodynamic instability, such as cyanosis, diaphoresis, altered mentation, pallor, and weak or thready pulses. Assess heart sounds for regularity and the lungs for signs of pulmonary congestion. In addition, assess lower extremities for edema. In patients who have altered mental status, palpation of the chest wall may reveal an implantable cardiac device, such as an automatic implantable cardioverter/defibrillator or a pacemaker. Dialysis catheters or arterial-venous fistulas or grafts also may be discovered by examination of the upper chest and extremities.
A 12-lead ECG is important in bradycardia evaluation and has the most diagnostic value, as it allows analysis of rhythm and potential ischemic changes. Although an ECG should be performed as soon as possible, obtaining an ECG should not delay the management of a hemodynamically unstable patient. Continuous cardiac monitoring while in the ED also will be helpful if the patient has changes in cardiac rhythm, hemodynamic status, or clinical presentation.
Serologic studies may be valuable in determining the underlying etiology of bradycardia. Quickly obtain serum electrolyte levels, such as potassium, and immediately address abnormalities if corresponding ECG findings are present. Blood urea nitrogen (BUN) and creatinine to assess for renal function and acute kidney injury also are essential. Serum cardiac markers, such as creatine kinase (CK) and troponin, as well as B-type natriuretic peptide (BNP) or NT-proBNP, will help to assess myocardial injury, ischemia, and heart failure, respectively. Thyroid function tests, such as TSH (thyroid stimulating hormone), may reveal underlying thyroid disease. In addition, serum drug levels for medications such as digoxin should be measured if the patient is taking the medication or if ingestion is suspected. Serologic titers for various infectious diseases such as Lyme disease may be helpful in a short-term capacity, but since the results will not be available immediately, they are not essential in the initial diagnostic workup.
Radiologic imaging may assist in diagnosis and management of underlying etiologies or resultant disease processes. A chest radiograph may reveal pulmonary vascular congestion (heart failure), pacemaker lead fracture (patients who have pacemakers that are failing to capture), or enlarged cardiac silhouette (possible pericardial effusion or cardiomegaly). Focused bedside ultrasound also is useful in assessing for pulmonary edema (presence of B-lines on lung exam), pericardial fluid, and left ventricular function.
Emergent cardiology consultation is prudent for all patients who are symptomatic or who have high-degree heart blocks, signs and/or symptoms of hypoperfusion or hemodynamic instability, ECG findings of AMI, or positive cardiac markers.
Differential Diagnosis
There are a few cardiac rhythms that potentially can be mistaken for a bradydysrhythmia, as the heart rate will be discordant to ECG findings. Ventricular bigeminy, blocked premature atrial contractions, and frequent premature ventricular contractions produce electrical impulses that will not manifest as contractions. Thus, the cardiac monitor or ECG will be discordant with the patient’s actual pulse rate, which may be bradycardic.
When evaluating patients who present to the ED with bradycardia, it is important for the clinician to consider the broad spectrum of diagnoses that may be responsible for this finding. It is helpful to think in terms of intrinsic and extrinsic causes, as outlined in the section on etiology. (See Table 1.) Although all of these potential causes will not be discussed, a few are worth mentioning.
Intrinsic
Cardiovascular disease, such as AMI or ischemia, should be suspected and identified expeditiously.
Infectious diseases may cause intrinsic injury to the nodal tissue via direct infiltration, leading to conduction abnormalities. Lyme disease is caused by the Borrelia burgdorferi bacteria and is transmitted via the Ixodes scapularis tick in the United States. Lyme disease may progress to carditis in up to 10% of untreated patients.14 It may affect any part of the heart, but the AV node is the most common location, resulting in a reversible AV block seen on ECG. Patients also may present with alternating or progressive AV conduction delays. Although there are no recent data, older studies have demonstrated that between 44% and 49% of patients with Lyme carditis experience third-degree AV block.15,16 Lyme carditis may occur in individuals of any age, but it should be suspected in younger patients with unexplained conduction abnormalities. Other infectious diseases, such as tertiary syphilis and viral illnesses (e.g., parvovirus and coxsackievirus), should be considered in patients who have unexplained cardiac findings.
Infiltrative diseases, such as sarcoidosis, also are in the differential diagnosis of bradycardia. Although primarily a pulmonary disease, sarcoidosis also can affect the heart and is associated with a poor prognosis. The most common arrhythmia in cardiac sarcoidosis is complete heart block, which has been reported in up to 30% of patients.17 Patients may present with heart block, syncope, or sudden death. Consider this diagnosis in patients without other etiologies of conduction disorders.
Extrinsic
Autonomic-mediated causes of bradydysrhythmias include carotid sinus stimulation or hypersensitivity, as well as vagal nerve stimulation induced by coughing, vomiting, defecation, or micturition.
Syncope may be a consequence of a cardiac arrhythmia. Syncope attributable to SA or AV dysfunction may be related to the poor physiologic adaptation from a drop in heart rate leading to poor perfusion.18
Chronotropic incompetence is a condition that involves an inadequate physiologic response in heart rate to exercise or physical activity, resulting in a “relative bradycardia.” Diagnosis of this phenomenon is difficult, and the parameters defined in the literature are varied and not validated. However, many clinicians consider chronotropic incompetence as failure to achieve 80% maximal heart rate (220 beats per minute minus age) at peak exercise.8
Abnormalities in electrolytes, such as potassium, calcium, and magnesium, must be considered, identified expeditiously, and treated in patients with ECG abnormalities. As the most abundant mineral in the body, calcium plays a vital role in many intracellular organ functions — particularly the heart. Hypercalcemia (defined as serum levels > 10.5 mg/dL) initially may increase contractility of the heart, but as levels exceed 15 mg/dL, myocardial depression may occur. Although the cardiovascular symptoms of hypercalcemia are variable, patients with total serum calcium levels > 15-20 mg/dL may develop AV block and cardiac arrest.19 Calcium has antagonistic effects on potassium and magnesium on a cellular level.
Elevated magnesium levels may cause cardiac arrhythmias, including bradycardia. Patients receiving magnesium for preeclampsia or asthma should be monitored or frequently assessed. Hyperkalemia or hypokalemia also may lead to severe cardiac manifestations. Hypokalemia may cause alterations in cardiac excitability and conduction. It should be noted that many patients who have hypercalcemia also have hypokalemia. Both conditions contribute to cardiac arrhythmias.19 As a special note, patients who take digitalis are particularly susceptible to the effects of hypokalemia and hypercalcemia, as they will have alterations in cardiac excitability and conduction.
Thyroid disease also should be considered. As a result of unclear pathophysiologic mechanisms, hypothyroid disease may manifest clinically in bradycardia and heart block. Bradycardia from hypothyroidism is likely reversible. Several case reports and studies demonstrate resolution of AV block with thyroid hormone replacement and normalization of the TSH in some patients.20,21 Although hyperthyroidism is associated more commonly with tachyarrhythmias, AV block secondary to thyrotoxicosis has been cited in the literature as a rare but potential cause.20,22
Hypothermia is defined as a core body temperature < 95°F (35°C) and may cause intraventricular conduction delays and bradydysrhythmias. An Osborn wave (also called a J wave) is a dome-shaped deflection in the same direction of the R wave and immediately follows the QRS complex. This characteristic ECG finding is present in 80% of patients with a core temperature below 30°C.23
Management
The approach to patients with bradycardia is based on the patient’s hemodynamic stability and presence or absence of symptoms. The management of hemodynamically stable patients who are asymptomatic depends on the rhythm and underlying etiology. Sinus bradycardia typically does not require intervention unless the heart rate is less than 50 beats per minute and the patient is symptomatic or shows signs of hypoperfusion.24
Patients with bradycardia who are hemodynamically unstable exhibit signs and symptoms of hypoperfusion, such as pallor, diaphoresis, altered mentation, cyanosis, chest pain, acute heart failure, and hypotension. Immediately place patients who have unstable vital signs or show signs of hypoperfusion on a cardiac monitor with continuous pulse oximetry monitoring. Obtain intravenous access and, if available, have the transcutaneous pacer at the patient’s bedside. Supply oxygen and perform aggressive airway and breathing management to patients in whom bradycardia is the suspected result of hypoxia.
The mainstays of medical treatment are atropine, dopamine, and epinephrine. Atropine is the first-line therapy for unstable bradycardia. Atropine is an anticholinergic agent that has a mechanism of action on cardiac activity via parasympathetic blockade and direct vagolytic action. This leads to an increased firing rate of the SA node and AV node conduction, thus increasing the heart rate. The initial dose of atropine is 0.5 mg IV bolus. This may be repeated every three to five minutes up to a maximum of 3 mg. In children, the dose is 0.02 mg/kg IV or IO with a minimum dose of 0.1 mg and a maximum of 0.5 mg. Caution should be taken in patients in whom AMI is suspected, as the increase in heart rate also may increase myocardial oxygen demand, leading to further ischemia or increase in the size of the infarction.24 Atropine has inconsistent success in patients with Mobitz type II AV block.25,26 Third-degree AV block secondary to infranodal pathology is unlikely to respond to atropine, as its mechanism of action is at the AV node. In addition, patients who have had cardiac transplantation no longer have innervation from the vagus nerve and, therefore, atropine likely will not be effective.27 However, there have been cases of AV block and sinus arrest following atropine use in heart transplant patients.27 If there is no improvement with atropine or if its maximum dose has been reached, either dopamine or epinephrine may be administered.
Dopamine and epinephrine both are catecholamines with alpha- and beta-adrenergic action and may be used as second-line treatments for unstable bradydysrhythmias. Intravenous dopamine infusion is initiated at a rate of 2 to 20 micrograms/kg/minute and titrated to the patient’s clinical response rather than a specific target heart rate. Epinephrine dosing is 2 to 10 micrograms/min or 0.1 to 0.5 mcg/kg/min and similarly is titrated to the patient’s hemodynamic and clinical response.
Isoproterenol is a beta-adrenergic agent with both beta-1 and beta-2 effects. The literature supporting the use of this drug is lacking and although it is not included in the American Heart Association Advanced Cardiac Life Support algorithm, its use may be considered.24 The recommended dose of isoproterenol is 2 to 10 mcg/min intravenously and is to be titrated based on clinical effect.
Bradydysrhythmias caused by overdose or toxicity of pharmacologic agents, either accidental or intentional, may have specific antidotes that will expedite management. For example, suspected digitalis toxicity may be treated with intravenous digoxin immune fragment antigen-binding. Unstable bradydysrhythmias secondary to beta-blockers may be treated with glucagon 5 mg intravenously slow bolus every 10 minutes, with a maximum of three doses. If the patient does not respond to two doses, he or she is unlikely to respond. If the patient does respond, an infusion of 2 to 5 mg/hour for adults can be started. Calcium channel blocker toxicity may be treated with intravenous calcium gluconate 10% or insulin. These medications may be considered in conjunction with recommendations for gastrointestinal decontamination in the appropriate circumstances. Poison centers may be helpful in guiding treatment in these difficult patients.
Cardiac Pacing
Zoll first introduced transcutaneous pacing (TCP) in 1952, and advancements in pacing technology over the years have made it the emergency pacing procedure of choice in multiple settings. TCP is a rapid, minimally invasive, and temporizing measure until transvenous pacing (TVP) can be implemented. TCP is indicated in patients with bradydysrhythmias who have hemodynamic instability or show signs of hypoperfusion and are not responsive to medical treatment. TCP should be considered in the management of patients who have high-degree AV blocks and bradydysrhythmias in the setting of AMI — particularly anterior/lateral MI.28 As stated in the previous section, it is prudent to have the TCP equipment at the bedside of patients with bradydysrhythmias so that there is no delay if pacing becomes necessary.
There are two self-adhesive pacer electrode pads: the anterior electrode (cathode, or negative electrode) and the posterior electrode. The anterior electrode should be placed on the left anterior chest wall, while the posterior electrode, which serves as the ground, may be placed either directly posterior to the anterior electrode on the patient’s back or on the lateral chest wall. Most electrodes have manufacturer instructions directly on the pads that direct appropriate placement. Recognizing that body habitus and grooming may be a challenge in electrode placement, it is important to provide optimal placement, as poor placement may interfere with capture.29 Remove transdermal patches prior to placement of the pacing electrodes. Shave excessive hair to ensure that the electrodes adhere appropriately to the patient. In women, place the anterior electrode beneath the breast, as breast tissue may cause transthoracic impedance.29 Take care to avoid placing pads over any implanted devices, such as a pacemaker or a defibrillator. Clinicians should be aware that there is a lack of standardization of equipment used by prehospital personnel and EDs. Thus, the pacing electrode pads placed on patients in the prehospital setting may not be compatible with equipment at the destination ED. Similarly, there may be differences in equipment between the ED and the patient’s inpatient disposition.
Once the pacing electrodes are on the patient and connected to the device, administer sedation to the patient, as the electrical impulses may be uncomfortable. Rate and current are adjustable. Start the rate at approximately 30 beats per minute above the patient’s inherent heart rate, keeping in mind that a heart rate between 60 and 70 beats per minute should be sufficient to achieve adequate perfusion. The current is measured in milliamperes (mA) and should be adjusted to the lowest setting that achieves capture while minimizing patient discomfort. This varies from individual to individual, and there are limited data regarding capture thresholds as they relate to pain tolerance. One study with 16 healthy male volunteers who were paced without sedation showed that the majority of subjects tolerated cardiac capture thresholds that ranged between 42 and 60 mA (mean 54 mA).30 A second study that compared multiple devices in only 10 subjects had capture thresholds ranging from 66.5 to 104 mA with variable pain thresholds. This suggests that a reasonable starting point would be at 50 mA with incremental increases of 5 to 10 mA until there is capture.
It is important to assess the patient for electrical and mechanical capture. Electrical capture is assessed on the ECG monitor. Mechanical capture may be determined by palpating the pulse at the carotid or femoral artery; however, the forceful external muscular contractions may make this challenging. Ultrasound has been useful in the assessment of capture.31-33 Once capture is achieved, set the current slightly above the capture threshold (5 to 10 mA). Failure to capture using TCP is an indication for TVP.
Chest compressions may be administered safely while the TCP is in use with little risk of injury to the healthcare provider — even directly over the pacer pads. This is because the power delivered with each impulse is minimal.34
Failure to recognize an underlying ventricular fibrillation, due to underlying pacing artifact, is the major potential complication of TCP. Other potential complications of TCP include pacer-induced ventricular fibrillation and cutaneous discomfort and injury.
In the ED setting, TVP is indicated for patients who have bradydysrhythmias in the setting of hemodynamic instability and are unresponsive to medication and TCP. As its name suggests, TVP is achieved by gaining central venous access and placing an electrode in the endocardium of the right ventricle. The most common emergency pacemaker code is VVI, which represents that the ventricle is paced, the ventricle is sensed, and there is inhibition of the pacemaker if a native impulse is sensed.
Although many of the specific steps involved in the actual TVP placement are beyond the scope of this review, important elements will be highlighted in this discussion.
Catheter site selection should be based on physician experience and comfort level and to leave options available for future permanent pacemaker placement. The options for central venous access in the setting of TVP are the internal jugular, subclavian, femoral, and brachial veins. The preferred locations are the right internal jugular and left subclavian veins, allowing the most direct anatomic routes to the right ventricle.35 Syverud et al found that these locations have higher rates of proper placement during cardiopulmonary arrest.34 Sterile preparation and technique, including guidance with ultrasound, for central venous access with TVP is the same as with usual central venous line placement.
The pacing catheter may be guided into proper location by ECG or ultrasound assistance. In using the ECG machine, the patient should be connected to the limb leads while the distal terminal (negative electrode) of the pacing catheter is placed on any of the V leads using an alligator clip. The other V leads should be attached as usual. As the pacing catheter is advanced, there will be characteristic ECG changes that correspond to its location. As ultrasound has become an essential diagnostic instrument in the ED, it offers the advantage of direct visualization of the pacer catheter in TVP placement as well as determination of mechanical capture.
Once the pacer catheter is against the right ventricular wall, the QRS segment will exhibit the typical ST-segment elevation, and the distal terminal may be connected to the corresponding terminal on the generator. Next, the pacer should be set to 80 beats per minute or 10 beats per minute above the patient’s underlying ventricular rate — whichever rate is faster. The generator then is set to full demand mode with an output of 5 mA and then slowly adjusted downward to achieve the minimal voltage required for capture. The generator is turned on, and the patient assessed for capture. The ideal threshold is less than 1.0 mA, and the final amperage should be 2.5 times that setting to ensure consistent capture. Perform a chest X-ray to confirm that the catheter tip is in the anterior-inferior aspect of the cardiac shadow. A chest radiograph also will rule out pneumothorax as a complication of central venous access.
Complications of TVP include the same complications that are encountered with central venous access. Additionally, there are complications that are associated with right heart catheterization, such as dysrhythmias including premature ventricular contractions and ventricular tachycardia, catheter misplacement, perforation of the ventricle or pericardium, and infection. Mechanical failure of the device also may occur.
Disposition
Determining the disposition patients who present with a bradydysrhythmia depends on several factors. Patients who present to the ED without symptoms and who are hemodynamically stable and remain asymptomatic throughout their ED course may be discharged home at the discretion of the emergency physician with outpatient follow-up. However, asymptomatic patients with a high-degree AVB, significant lab abnormalities, or other conditions may warrant admission or observation.
Symptomatic patients warrant admission with telemetry or observation in a monitored setting based on ECG findings, comorbid illnesses, laboratory findings, and underlying etiology. Patients who exhibit signs of hypoperfusion or hemodynamic instability or who have signs and/or symptoms of cardiac ischemia or infarction warrant admission to an intensive care setting. The emergency physician also must consider the availability of cardiology consultants in his or her facility, as well as the facility’s capability to care for the critically ill patient. Transfer to a facility with cardiac specialty services is indicated if the presenting facility cannot provide these services if required.
REFERENCES
- Spodick DH, Raju P, Bishop RL, Rifkin RD. Operational definition of normal sinus heart rate. Am J Cardiol 1992;69:1245-1246.
- Matelot D, Schnell F, Kervio G, et al. Athlete’s bradycardia may be a multifactorial mechanism. J Appl Physiol (1985) 2013;114:1755-1756.
- Pejkovic B, Krajnc I, Anderhuber F, Kosutic D. Anatomical aspects of the arterial blood supply to the sinoatrial and atrioventricular nodes of the human heart. J Int Med Res 2008;36:691-698.
- Saremi F, Abolhoda A, Ashikyan O, et al. Arterial supply to sinuatrial and atrioventricular nodes: Imaging with multidetector CT. Radiology 2008;246:99-107; discussion 8-9.
- James TN. The coronary circulation and conduction system in acute myocardial infarction. Prog Cardiovasc Dis 1968;10:410-449.
- Dobrzynski H, Boyett MR, Anderson RH. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation 2007;115:1921-1932.
- Lamas GA, Lee K, Sweeney M, et al. The mode selection trial (MOST) in sinus node dysfunction: Design, rationale, and baseline characteristics of the first 1000 patients. Am Heart J 2000;140:541-551.
- Epstein AE, DiMarco JP, Ellenbogen KA, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2013;61:e6-75.
- Cheng S, Keyes MJ, Larson MG, et al. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA 2009;301:2571-2577.
- Crisel RK, Farzaneh-Far R, Na B, Whooley MA. First-degree atrioventricular block is associated with heart failure and death in persons with stable coronary artery disease: Data from the Heart and Soul Study. Eur Heart J 2011;32:1875-1880.
- Nikolaidou T, Ghosh JM, Clark AL. Outcomes related to first-degree atrioventricular block and therapeutic implications in patients with heart failure. JACC: Clinical Electrophysiology 2016;2:181-192.
- Trappe HJ. Tachyarrhythmias, bradyarrhythmias and acute coronary syndromes. J Emerg Trauma Shock 2010;3:137-142.
- Berger PB, Ryan TJ. Inferior myocardial infarction. High-risk subgroups. Circulation 1990;81:401-411.
- Fish AE, Pride YB, Pinto DS. Lyme carditis. Infect Dis Clin North Am 2008;22:275-288, vi.
- McAlister HF, Klementowicz PT, Andrews C, et al. Lyme carditis: An important cause of reversible heart block. Ann Intern Med 1989;110:
339-345. - van der Linde MR. Lyme carditis: Clinical characteristics of 105 cases. Scand J Infect Dis Suppl 1991;77:81-84.
- Sekhri V, Sanal S, Delorenzo LJ, et al. Cardiac sarcoidosis: A comprehensive review. Arch Med Sci 2011;7:546-554.
- Saklani P, Krahn A, Klein G. Syncope. Circulation 2013;127:1330-1339.
- ECC Committee Task Forces of the American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 10.1: Life-Threatening Electrolyte Abnormalities. Circulation 2005;112(24suppl):IV121-125.
- Ozcan KS, Osmonov D, Erdinler I, et al. Atrioventricular block in patients with thyroid dysfunction: Prognosis after treatment with hormone supplementation or antithyroid medication. J Cardiol 2012;60:327-332.
- Schoenmakers N, de Graaff WE, Peters RH. Hypothyroidism as the cause of atrioventricular block in an elderly patient. Neth Heart J 2008;16:57-59.
- Topaloglu S, Topaloglu OY, Ozdemir O, et al. Hyperthyroidism and complete atrioventricular block — a report of 2 cases with electrophysiologic assessment. Angiology 2005;56:217-220.
- Alhaddad IA, Khalil M, Brown EJ, Jr. Osborn waves of hypothermia. Circulation 2000;101:E233-E244.
- Neumar RW, Otto CW, Link MS, et al. Part 8: Adult advanced cardiovascular life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010;122:S729-S767.
- Brady WJ, Swart G, DeBehnke DJ, et al. The efficacy of atropine in the treatment of hemodynamically unstable bradycardia and atrioventricular block: Prehospital and emergency department considerations. Resuscitation 1999;41:47-55.
- Swart G, Brady WJ Jr, DeBehnke DJ, et al. Acute myocardial infarction complicated by hemodynamically unstable bradyarrhythmia: Prehospital and ED treatment with atropine. Am J Emerg Med 1999;17:647-652.
- Bernheim A, Fatio R, Kiowski W, et al. Atropine often results in complete atrioventricular block or sinus arrest after cardiac transplantation: An unpredictable and dose-independent phenomenon. Transplantation 2004;77:1181-1185.
- O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013;127:529-555.
- Deakin CD, Nolan JP, European Resuscitation Council. European Resuscitation Council guidelines for resuscitation 2005. Section 3. Electrical therapies: Automated external defibrillators, defibrillation, cardioversion and pacing. Resuscitation 2005;67Suppl1:S25-S37.
- Falk RH, Zoll PM, Zoll RH. Safety and efficacy of noninvasive cardiac pacing. A preliminary report. N Engl J Med 1983;309:1166-1168.
- Ettin D, Cook T. Using ultrasound to determine external pacer capture. J Emerg Med 1999;17:1007-1009.
- Holger JS, Lamon RP, Minnegan HJ, Gornick CC. Use of ultrasound to determine ventricular capture in transcutaneous pacing. Am J Emerg Med 2003;21:227-229.
- Holger JS, Minnigan HJ, Lamon RP, Gornick CC. The utility of ultrasound to determine ventricular capture in external cardiac pacing. Am J Emerg Med 2001;19:134-136.
- Syverud SA, Dalsey WC, Hedges JR. Transcutaneous cardiac pacing. Ann Emerg Med 1984;13:982.
- Bessman ES. Emergency Cardiac Pacing. Roberts and Hedges’ Clinical Procedures in Emergency Medicine, 6th ed. Philadephia, PA: Elsevier Saunders; 2013:277-297.
- Mangrum JM, DiMarco JP. The evaluation and management of bradycardia. N Engl J Med 2000;342:703-709.
In this article, the normal anatomy and pathophysiology of bradydysrhythmias in adults will be reviewed, followed by the latest recommendations in evaluation and management.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.