Acute Kidney Injury in Patients With Cirrhosis
By Arnaldo Lopez Ruiz, MD, and Alexander S. Niven, MD
Dr. Lopez Ruiz is Attending Physician, Division of Critical Care, AdventHealth Medical Group, AdventHealth Orlando, FL. Dr. Niven is Senior Associate Consultant, Division of Pulmonary/Critical Care Medicine, Mayo Clinic, Rochester, MN.
Dr. Lopez Ruiz and Dr. Niven report no financial relationships relevant to this field of study.
Acute kidney injury (AKI) is a common clinical problem in the critically ill, associated with a two- to six-fold increased risk of death depending on its severity.1 Patients hospitalized with cirrhosis are a particularly high-risk population for this complication, often with devastating results. Nearly 30% of these patients develop AKI, with reported mortality rates as high as 50% to 90%.2
More than 60% of AKI cases in patients with cirrhosis are attributable to prerenal factors, such as hypovolemia, hypotension, or hypoperfusion.2 Hepatorenal syndrome (HRS) is a distinct form of prerenal kidney injury caused by the pathophysiologic abnormalities of systemic arterial vasodilatation and renal vasoconstriction found in cirrhosis. Patients with cirrhosis and ascites have a 50% probability of developing HRS within a five-year period, and HRS has been reported in approximately 20% of patients with cirrhosis and with AKI.3 Recent reports have shown the presence of urinary biomarkers for tubular injury and cases of acute tubular necrosis (ATN) in HRS, suggesting that this condition represents a continuous spectrum that starts with functional abnormalities and may progress to include intrinsic renal damage.4
Serum creatinine (sCr) remains the most practical biomarker of renal function in patients who have AKI with or without cirrhosis, although its limitations are more pronounced in patients with liver disease. Malnutrition, muscle wasting, and reduced liver function in patients with advanced cirrhosis can reduce sCr formation by more than 20%, while tubular secretion is decreased. The increased volume of distribution often seen in cirrhosis may dilute sCr, and some laboratory assays (Jaffe reaction-based) give a falsely low sCr in the setting of high bilirubin levels (> 2 mg/dL). These effects often result in an overestimate of glomerular filtration rate (GFR) in cirrhotic patients using sCr alone.5 Previous criteria for the diagnosis of AKI or HRS in cirrhosis using a fixed threshold of sCr (≥ 1.5 mg/dL for AKI or ≥ 2.5 mg/dL for HRS)6,7 are therefore problematic. An sCr ≥ 1.5 mg/dL in this population often signifies a marked reduction in GFR (≤ 25 mL/min), and these fixed thresholds do not consider dynamic sCr changes in the preceding days or weeks necessary to distinguish between acute and chronic kidney injury.5
PATHOPHYSIOLOGY
The pathophysiology of HRS is complex. Current evidence suggests there are two closely interrelated mechanisms contributing to the renal dysfunction in cirrhosis. Elevated portal pressure causes endothelial sheer stress in the splanchnic circulation, resulting in increased synthesis of nitric oxide, which contributes to peripheral arterial vasodilation in the splanchnic and systemic circulation. This vasodilatory state leads to renal hypoperfusion and subsequent activation of the renin-angiotensin-aldosterone system (RAAS), arginine-vasopressin, and endothelin-1 that results in further renal vasoconstriction and persistent hypoperfusion. RAAS activation also increases sodium-water retention, ascites, and peripheral edema formation, and increases sympathetic activity, which, combined with systemic vasodilatation, contributes to high cardiac output and hyperdynamic circulation.8-10 Patients with cirrhosis also have evidence of increased systemic inflammation, with elevated levels of tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), interferon-gamma (INF-γ), C-reactive protein (CRP), and reactive oxygen species in > 80% of these patients.11-13 These findings have been attributed to increased translocation of viable bacteria or bacterial products (pathogen-associated molecular patterns, or PAMPs) from the intestinal lumen to the mesenteric lymph nodes and then to the intrahepatic reticuloendothelial system as a consequence of intestinal bacterial overgrowth, structural abnormalities in the intestinal mucosa, and reduced intestinal mucosal immune function that commonly are present in patients with cirrhosis.8,14 This inflammatory cascade contributes to arterial vasodilatation through further nitric oxide synthesis and also can cause direct tissue damage, evidenced by increased levels of urinary biomarkers of tubular injury (β2-microglobulin or neutrophil-gelatinase associated lipocalin [NGAL]) and features of ATN that have been identified now using electron microscopy in renal biopsies of patients with AKI-HRS.4,15
PRECIPITATING FACTORS
HRS develops in clinical situations that exacerbate the underlying vasodilatation and systemic inflammation in patients with cirrhosis. Bacterial infection is considered the most common precipitant of HRS. In patients listed for liver transplantation, bacterial infection due to urinary tract infections, cellulitis, spontaneous bacterial peritonitis (SBP), and bacteremia were the precipitating factors for 68% to 75% of HRS cases.16 SBP is accepted as the most common precipitating event for HRS.17 In fact, SBP prophylaxis is associated with a reduction in the subsequent development of HRS among patients with cirrhosis.18
Diuretic therapy at high doses can further exaggerate the reduction in effective arterial blood volume present in advanced cirrhosis and contribute to the development of HRS. Acute blood loss from gastrointestinal bleeding (GIB) also can reduce effective arterial blood volume and increase renal vasoconstriction, worsening renal perfusion.8 Many cirrhosis patients with GIB release multiple pro-inflammatory cytokines due to stress and bacteremia that further predispose them to HRS.9
Removal of more than 5 L (≥ 3 L in patients with body mass index [BMI] ≤ 20) of ascitic fluid can reduce intra-abdominal pressure significantly, increasing venous return and exaggerating splanchnic vasodilatation within the first 24 hours. These changes trigger the activation of various vasoconstrictor systems over the following week, a phenomenon known as post-paracentesis circulatory dysfunction.9,19 Renal dysfunction can occur in 20% of patients after large volume paracentesis and can be prevented in about 60% of cases by using intravenous hyperoncotic colloid (albumin 25% – 8 g per liter of fluid removed in this setting).19
Acute-on-chronic liver failure due to conditions such as alcoholic hepatitis or ischemic liver injury often is associated with substantial intrahepatic inflammation, increasing the risk for HRS.20 In a well-designed meta-analysis, neither corticosteroids nor pentoxifylline has been shown to reduce the incidence of HRS during acute alcoholic hepatitis in patients with underlying alcoholic cirrhosis.21 HRS in patients with acute-on-chronic liver failure is more likely to have evidence of structural renal damage and, therefore, is more often prolonged and associated with more severe stages of renal dysfunction than other causes of renal failure in cirrhosis.22
DIAGNOSIS
New guidelines proposed by the International Ascites Club (IAC) and the Acute Dialysis Quality Initiative Group define AKI in cirrhosis as an increase in sCr by 0.3 mg/dL in less than 48 hours or an increase of ≥ 50% from baseline stable sCr within the previous three months.23 This revised definition allows for AKI diagnosis at an earlier stage of renal dysfunction, facilitating earlier therapeutic intervention and identification of AKI cases (stages 2 and 3) usually associated with major complications (volume overload, acid-base imbalance, or electrolyte abnormalities) that may benefit from renal replacement therapy.
HRS is defined as the development of renal failure in patients who have advanced liver failure (acute or chronic) in the absence of identifiable causes of renal pathology. It is divided into two types: acute, or type 1 HRS, and chronic, or type 2 HRS. New guidelines recognize type 1 HRS as acute kidney injury–hepatorenal syndrome (AKI-HRS) with a doubling of sCr from baseline without the prior rigid threshold of sCr 2.5 mg/dL or two-week time frame previously required for this diagnosis.10,23 In the same manner, type 2 HRS now is considered a form of chronic kidney disease (CKD-HRS) in which there is a steady increase in sCr over a period of months, usually in a patient with advanced cirrhosis (MELD > 20) and refractory ascites.23 Low urinary sodium (< 20 mmol/L), low fractional excretion of sodium (FENa < 1%), and low fractional excretion of urea (FEUrea < 28%) are sensitive measurements that have been found consistently in AKI-HRS, but with very low specificity for this condition.24 A previous large, multicenter observational study of urinary biomarkers of AKI in cirrhosis has shown that FENa was the only biomarker to distinguish HRS from prerenal azotemia when a cut-off < 0.1% is used.15 In addition, FEUrea > 28% also is valuable for distinguishing HRS from ATN, particularly in the presence of diuretics.25 Serum cystatin C correlates with GFR better than sCr and has been proposed to be a more accurate marker of kidney function in cirrhosis. In patients with cirrhosis, cystatin C has been shown to be an independent predictor for HRS and one-year survival.12,13 However, cystatin C assays are not uniformly available in clinical laboratories, and the practicality of implementing their use is questionable.
TREATMENT OF AKI IN CIRRHOSIS
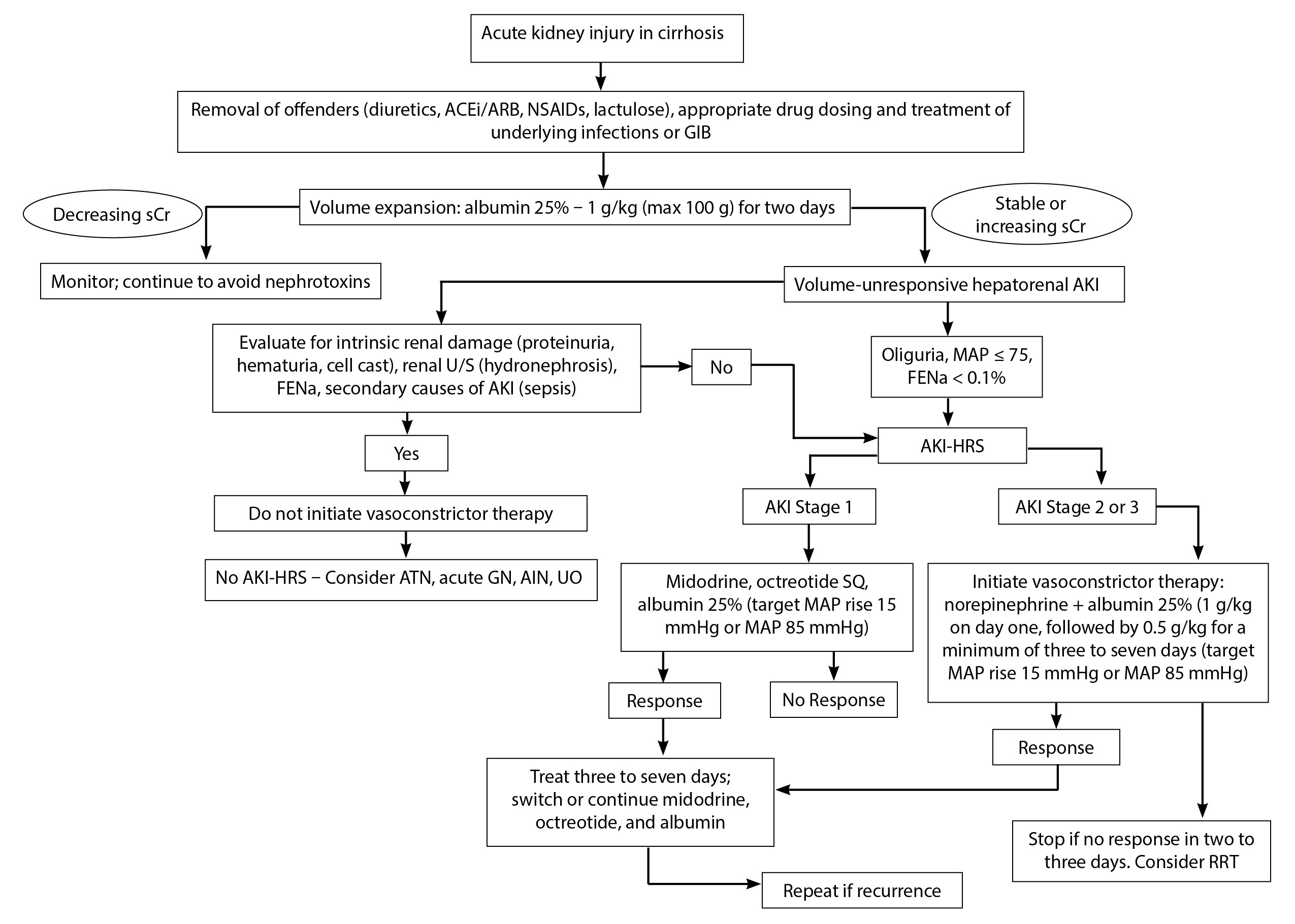
Early recognition is essential to effectively managing AKI in cirrhosis patients. (See Figure 1.) Current guidelines recommend systematic evaluation of bacterial infection by culturing all possible sites (blood, urine, ascites, sputum) and inspecting the skin surfaces for cellulitis. The threshold for starting antibiotics should be low if clinical concern for infection is present. Administration of hyperoncotic albumin with a broad-spectrum cephalosporin in patients with SBP prevents AKI-HRS.26 Since HRS is a diagnosis of exclusion, urine should be examined for proteinuria, hematuria, and casts to evaluate for other causes of parenchymal renal disease. Nephrotoxic exposures (radiographic dyes, nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers) should be minimized or withdrawn, and diuretics often are held in this setting.23 Acute blood loss anemia due to GIB should be corrected to a hemoglobin of 7-8 g/dL, since this restrictive approach has been shown to reduce the risk for further bleeding in patients with cirrhosis.27
Figure 1: Algorithm for the Management of Acute Kidney Injury-Hepatorenal Syndrome |
 |
|
ATN = acute tubular necrosis; AIN = acute interstitial nephrosis; Acute GN = acute glomerulonephritis; UO = obstructive uropathy |
Patients with cirrhosis are at high risk of prerenal azotemia, and guidelines recommend a trial of albumin 25% at a dose of 1 g/kg of actual body weight up to a maximum dose of 100 g/day for at least 48 hours in the absence of other complicating conditions.10,23 AKI often will improve once precipitating events are removed and volume replacement is administered. Patients whose renal function does not return to baseline are considered to have a “volume nonresponsive AKI.”
Patients with “volume nonresponsive AKI” are at high risk for AKI-HRS, especially when evidence of renal hypoperfusion (oliguria, FENa < 0.1%) is present. These patients should receive vasopressor therapy to correct hypotension or improve mean arterial pressure (MAP) combined with hyperoncotic albumin administration.23 A pooled analysis of data from 501 patients across 21 studies showed a strong direct correlation between the magnitude of rise in MAP and the improvement in renal function (absolute decrease in sCr) during vasoconstrictor therapy, irrespective of the vasoconstrictor used.28 Contrary to common assumption, the MAP in patients with AKI-HRS is approximately 70 mmHg in published studies. Targeting a MAP of 85-90 mmHg may not be unreasonable, and it was associated with greater chances of renal recovery (reversal of AKI-HRS), less requirement of dialysis, and better short-term and long-term overall survival.29,30 Because of the lack of availability of the vasopressin analog terlipressin in the United States, the preferred vasoconstrictor agent is norepinephrine. (See Figure 1.) Multiple controlled studies have demonstrated that norepinephrine was as effective as terlipressin, reversing AKI-HRS in 45-60% of cases. Compared to those treated with terlipressin, patients treated with norepinephrine had a similar 30-day mortality (40-48%), similar 15-day relapse rates, but with significantly less severe ischemic complications.31-33 The recommended duration of vasoconstrictor therapy is a minimum of three days, and therapy should be extended to seven days in those patients who show favorable response.8,16,23,34 Because of the high rate of recurrence (40-60%) of AKI-HRS within 15 days, patients who experience a decline in renal function after stopping vasoconstrictor therapy should be re-treated with the same protocol.8,23
Because of the logistics and expense related with norepinephrine administration, the most widely used therapy to treat AKI-HRS is the combination of oral midodrine (α1 agonist), subcutaneous octreotide (somatostatin analog), and albumin. Many small prospective and retrospective studies have compared this combination to midodrine or albumin and showed mild benefits in renal function.35 However, it is important to recognize that in comparison studies, norepinephrine has been shown to be more effective than this combination to consistently increase MAP, improve AKI-HRS, and reduce its recurrence.36
The contribution of portal hypertension in AKI-HRS development has prompted consideration of trans-jugular intrahepatic portosystemic shunt (TIPS) as a therapy for this condition.37 Small-scale studies of TIPS in advanced cirrhosis have reported an improvement in kidney function at 30 days, reduced need for dialysis, and improved survival.38,39 A systematic review of nine studies showed a pooled rate of improvement in kidney function of 93%, but a 46% rate of hepatic encephalopathy in addition to common periprocedural complications.40
The definitive treatment for AKI-HRS is liver transplantation. It eliminates portal hypertension and liver dysfunction, the two pivotal pathogenetic mechanisms for the development of AKI-HRS. An estimated 65-75% of patients experience resolution of AKI-HRS after liver transplantation,41 with lower recovery for patients on dialysis at the time of transplantation.42 The lack of recovery in renal function is most likely secondary to structural renal damage induced by prolonged or unrecovered AKI-HRS.42 For patients who are eligible for liver transplantation, vasoconstrictor therapy is viewed as “bridge” therapy until a suitable donor is found. Among patients who are not candidates for liver transplantation, short-term survival is increased in those who respond to vasoconstrictor therapy.43,44 In a controlled study using vasoconstrictor therapy, the three-month survival rate of patients who achieved resolution of AKI-HRS was 40% compared to 4% for those who did not respond to treatment.43 Therefore, patients who have AKI-HRS should receive a timely liver transplant, especially in those who do not respond to vasoconstrictor therapy. The United Network for Organ Sharing in the United States has recommended that patients with AKI-HRS who have had dialysis for more than eight weeks in the pretransplant period be considered for a combined liver-kidney transplant (CLKT), indicating the unlikely event of reversal of renal dysfunction with liver transplant alone.45
SUMMARY
AKI and HRS are common conditions found in patients with cirrhosis, due to the systemic vasodilation and systemic inflammation frequently found in this population. HRS is recognized now to be a clinical continuum, starting with functional renal hypoperfusion, but often progressing to acute tubular injury if left untreated. As a result, guidelines have been revised recently to facilitate earlier recognition of these conditions, and aggressive management that includes systematic identification and treatment of precipitating factors, volume expansion with hyperoncotic albumin, and vasopressor administration to restore renal perfusion and reduce the risk of parenchymal damage. TIPS may be considered in refractory cases of AKI-HRS, but it is associated with a high rate of encephalopathy in this population, and early liver transplantation offers definitive treatment when performed early.
REFERENCES
- Singbartl K, Kellum JA. AKI in the ICU: Definition, epidemiology, risk stratification, and outcomes. Kidney Int 2012;81:819-825.
- Garcia-Tsao G, Parikh CR, Viola A. Acute kidney injury in cirrhosis. Hepatology 2008;48:2064-2077.
- Allegretti AS, Ortiz G, Wenger J, et al. Prognosis of acute kidney injury and hepatorenal syndrome in patients with cirrhosis: A prospective cohort study. Int J Nephrol 2015;2015:108139.
- Adebayo D, Morabito V, Davenport A, Jalan R. Renal dysfunction in cirrhosis is not just a vasomotor nephropathy. Kidney Int 2015;87:509.
- Sherman DS, Fish DN, Teitelbaum I. Assessing renal function in cirrhotic patients: Problems and pitfalls. Am J Kidney Dis 2003;41:269-278.
- Arroyo V, Ginès P, Gerbes AL, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. Hepatology 1996;23:164-176.
- Bataller R, Ginès P, Guevara M, Arroyo V. Hepatorenal syndrome. Semin Liver Dis 1997;17:233-247.
- Angeli P, Morando F. Optimal management of hepatorenal syndrome in patients with cirrhosis. Hepat Med 2010;21:87.
- Bernardi M, Moreau R, Angeli P, et al. Mechanisms of decompensation and organ failure in cirrhosis: From peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol 2015;63:1272.
- Wong F. The evolving concept of acute kidney injury in patients with cirrhosis. Nat Rev Gastroenterol Hepatol 2015;12:711.
- Geetha A, Lakshmi Priya MD, Jeyachristy SA, Surendran R. Level of oxidative stress in the red blood cells of patients with liver cirrhosis. Indian J Med Res 2007;126:204-210.
- Gomaa SH, Shamseya MM, Madkour MA. Clinical utility of urinary neutrophil gelatinase-associated lipocalin and serum cystatin C in a cohort of liver cirrhosis patients with renal dysfunction: A challenge in the diagnosis of hepatorenal syndrome. Eur J Gastroenterol Hepatol 2019;31:692-702.
- Ahn HS, Kim YS, Kim SG, et al. Cystatin C is a good predictor of hepatorenal syndrome and survival in patients with cirrhosis who have normal serum creatinine levels. Hepatogastroenterology 2012;59:1168-1173.
- Dirchwolf M, Ruf AE. Role of systemic inflammation in cirrhosis: From pathogenesis to prognosis. World J Hepatol 2015;7:1974-1981.
- Belcher JM, Sanyal AJ, Peixoto AJ, et al; TRIBE-AKI Consortium. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology 2014;60:622-632.
- Salerno F, Gerbes A, Ginès P, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007;56:1310-1318.
- Hampel H, Bynum GD, Zamora E, El-Serag HB. Risk factors for the development of renal dysfunction in hospitalized patients with cirrhosis. Am J Gastroenterol 2001;96:2206-2210.
- Fernandez J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology 2007;133:818-824.
- European Association for the Study of the Liver; Gines P, Angeli P, Lenz K, et al. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol 2010;53:397-417.
- Davenport A, Sheikh MF, Lamb E, et al. Acute kidney injury in acute-on-chronic liver failure: Where does hepatorenal syndrome fit? Kidney Int 2007;92:1058-1070.
- Njei B, Do A, McCarty TR, Fortune BE. Corticosteroids versus pentoxifylline for severe alcoholic hepatitis: A sequential analysis of randomized controlled trials. J Clin Gastroenterol 2016;50:871-881.
- Arora R, Kathuria S, Jalandhara N. Acute renal dysfunction in patients with alcoholic hepatitis. World J Hepatol 2011;3:121-124.
- Angeli P, Gines P, Wong F, et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: Revised consensus recommendations of the International Club of Ascites. Gut 2015;64:531.
- Alsaad AA, Wadei HM. Fractional excretion of sodium in hepatorenal syndrome: Clinical and pathological correlation. World J Hepatol 2016;8:1497-1501.
- Patidar KR, Kang L, Bajaj JS, et al. Fractional excretion of urea: A simple tool for the differential diagnosis of acute kidney injury in cirrhosis. Hepatology 2018;68:224-233.
- Sort P, Navasa M, Arroyo V, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med 1999;341:403-409.
- Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11-21.
- Velez JC, Nietert PJ. Therapeutic response to vasoconstrictors in hepatorenal syndrome parallels increase in mean arterial pressure: A pooled analysis of clinical trials. Am J Kidney Dis 2011;58:928-938.
- Maddukuri G, Cai CX, Munigala S, et al. Targeting an early and substantial increase in mean arterial pressure is critical in the management of type 1 hepatorenal syndrome: A combined retrospective and pilot study. Dig Dis Sci 2014;59:471-448.
- Velez JC, Kadian M, Taburyanskaya M, et al. Hepatorenal acute kidney injury and the importance of raising mean arterial pressure. Nephron 2015;131:191-201.
- Singh V, Ghosh S, Singh B, et al. Noradrenaline vs. terlipressin in the treatment of hepatorenal syndrome: A randomized study. J Hepatol 2012;56:1293-1298.
- Alessandria C, Ottobrelli A, Debemardi-Venon W, et al. Noradrenalin vs terlipressin in patients with hepatorenal syndrome: A prospective, randomized, unblinded, pilot study. J Hepatol 2007;47:499-505.
- Saif RU, Dar HA, Sofi SM, et al. Noradrenaline versus terlipressin in the management of type 1 hepatorenal syndrome: A randomized controlled study. Indian J Gastroenterol 2018;37:424-429.
- Wong F, Angeli P. New diagnostic criteria and management of acute kidney injury. J Hepatol 2017;66:860.
- Angeli P, Volpin R, Gerunda G, et al. Reversal of type 1 hepatorenal syndrome with the administration of midodrine and octreotide. Hepatology 1999;29:1690-1697.
- Tavakkoli H, Yazdanpanah K, Mansourian M. Noradrenaline versus the combination of midodrine and octreotide in patients with hepatorenal syndrome: Randomized clinical trial. Int J Prev Med 2012;3:764-769.
- Guevara M, Gines P, Bandi JC, et al. Transjugular intrahepatic portosystemic shunt in hepatorenal syndrome: Effects on renal function and vasoactive systems. Hepatology 1998;28:416-422.
- Brensing KA, Textor J, Perz J, et al. Long term outcome after transjugular intrahepatic portosystemic stent-shunt in non-transplant cirrhotics with hepatorenal syndrome: A phase II study. Gut 2000;47:288-295.
- Anderson CL, Saad WE, Kalagher SD, et al. Effect of transjugular intrahepatic portosystemic shunt placement on renal function: A 7-year, single-center experience. J Vasc Interv Radiol 2010;21:1370-1376.
- Song T, Rossle M, He F, et al. Transjugular intrahepatic portosystemic shunt for hepatorenal syndrome: A systematic review and meta-analysis. Dig Liver Dis 2018;50:323-330.
- Wong F, Leung W, Al Beshir M, et al. Outcomes of patients with cirrhosis and hepatorenal syndrome type 1 treated with liver transplantation. Liver Transpl 2015;21:300-307.
- Sharma P, Goodrich NP, Zhang M, et al. Short- term pretransplant renal replacement therapy and renal nonrecovery after liver transplantation alone. Clin J Am Soc Nephrol 2013;8:1135-1142.
- Boyer TD, Sanyal AJ, Garcia-Tsao G, et al. Impact of liver transplantation on the survival of patients treated for hepatorenal syndrome type 1. Liver Transpl 2011;17:1328-1332.
- Heidemann J, Bartels C, Berssenbrugge C, et al. Hepatorenal syndrome: Outcome of response to therapy and predictors of survival. Gastroenterol Res Pract 2015;2015:457613.
- Hussain SM, Sureshkumar KK. Refining the role of simultaneous liver kidney transplantation. J Clin Transl Hepatol 2018;6:289-295.
Acute kidney injury and hepatorenal syndrome are common conditions found in patients with cirrhosis, due to the systemic vasodilation and systemic inflammation frequently found in this population.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.