Diabetes in Pediatric ED Patients
AUTHORS
Taryn Taylor, MD, MEd, FAAP, FACEP, Assistant Professor of Pediatrics and Emergency Medicine, Emory University School of Medicine, Atlanta
Sherita Holmes, MD, Assistant Professor of Pediatrics and Emergency Medicine, Emory University School of Medicine, Atlanta
PEER REVIEWER
Steven M. Winograd, MD, FACEP, Mt. Sinai Queens Hospital Center, Jamaica Queens, NY; Assistant Clinical Professor of Emergency Medicine, Mt. Sinai Medical School, New York City; Assistant Clinical Professor of Emergency Medicine, NYiTCOM, Old Westbury, NY
Emergency medicine providers commonly will encounter children with type 1 and type 2 diabetes. Unfortunately, the incidence of both is increasing, and the acute care provider must be able to recognize the subtle and dramatic presentations of both diseases. Early recognition and management of both the disease and its complications — diabetic ketoacidosis, hyperglycemic hyperosmolar state, and cerebral edema — are critical to ensure an optimal outcome.
— Ann Dietrich, Editor, MD, FAAP, FACEP
Definition
Diabetes mellitus (DM) is a metabolic condition in which the body has elevated blood glucose levels that, if left untreated, can lead to significant damage to multiple organ systems, including the kidneys, eyes, heart, vasculature, skin, and brain. Two common forms of DM afflict children and adolescents. Type 1 DM (T1 DM) is the most common. However, because of the obesity epidemic, type 2 DM (T2 DM) is becoming increasingly more prevalent.1
T1 DM stems from a lack of insulin production due to autoimmune destruction of the endocrine cells of the pancreas.2 Because of this lack of insulin production, the body goes into ketosis and hyperglycemia. If left untreated, this progresses to diabetic ketoacidosis (DKA). In contrast, in T2 DM, the body cannot use insulin properly because of insulin resistance. Regardless of the etiology, the inability of the body to use insulin leads to derangements in the metabolism of fats, carbohydrates, and proteins.3
Epidemiology
Both types of diabetes are increasing in the United States. T1 DM represents one of the most common chronic pediatric conditions in most western countries, accounting for approximately 90% pediatric cases of diabetes.3 In contrast, T1 DM accounts for only 5% to 10% of adults with diabetes.4 Between 2002 and 2012, the incidence of T1 DM increased by an estimated 1.8% annually in the United States.5 Furthermore, when analyzing the incidence among racial and ethnic groups, there are high relative increased rates of T1 and T2 DM in other groups vs. non-Hispanic whites.5
Etiology
T1 DM is a disease characterized by a relative insulin deficiency caused by beta cell destruction of the endocrine pancreas, also called islets of Langerhans. (See Figure 1.) This destruction is a chronic autoimmune-mediated process. Once approximately 90% of pancreatic beta cells are destroyed, patients exhibit clinical symptoms.3 The etiology of T1 DM is multifactorial — environmental factors, genetic predisposition, and the immune system all contribute to its development.6 Multiple genes lead to T1 DM susceptibility. High-risk human leukocyte antigen (HLA) genotypes have been linked explicitly to 40% to 50% of the genetic risk for progression to T1 DM.2 There is an increased lifetime risk of developing T1 DM in close relatives of patients diagnosed with T1 DM. Having multiple family members with T1 DM enhances this risk.7
Figure 1. Diagram of the Pancreas and Islets of Langerhans |
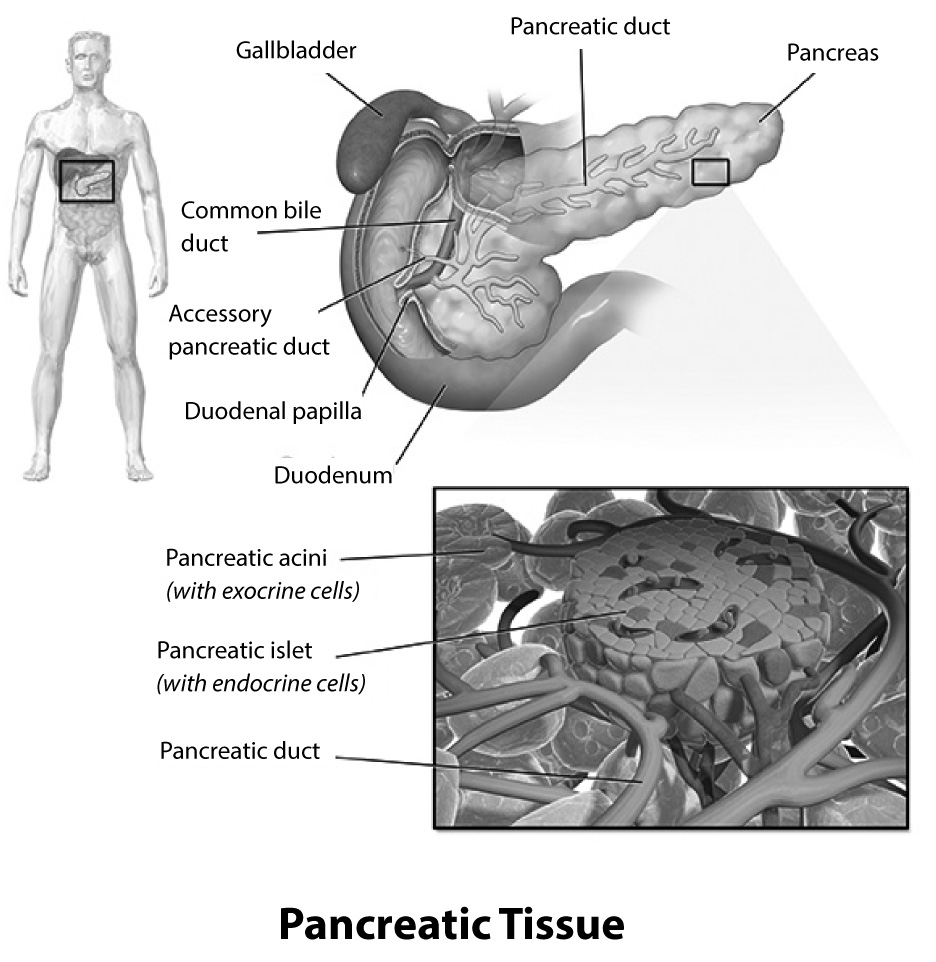 |
|
Image courtesy of: Blausen.com staff (2014). "Medical gallery of Blausen Medical 2014." WikiJournal of Medicine 1(2). doi:10.15347/wjm/2014.010. ISSN 2002-4436. |
Regarding environmental factors, viruses are presumed to be the most common triggers, particularly enterovirus. Studies have found a clinically significant association of enterovirus to T1 DM.2,6,8 Possible theories are that the virus directly kills the beta cells, or that it causes an exaggerated inflammatory response, leading to beta cell destruction by autoreactive T-cells.6 Other theories postulate that enterovirus triggers the first appearance of the autoantibody or speeds up the progression to clinical onset.7 Congenital rubella also has been associated to T1 DM, but there are no data linking other viruses, such as influenza.3 These are merely associations, and a direct causal link has not been proven.
The primary autoantibodies detected in patients with T1 DM are glutamic acid decarboxylase (GAD), insulinoma-associated-2 (IA2) or tyrosine phosphatase-2, insulin (IAA), and zinc transporter-8 (ZnT8). One or more islet autoantibodies were present in more than 90% of individuals diagnosed with T1 DM.3,9,10 The latency period from when autoantibodies are first present to clinical symptoms can take weeks to decades. Ziegler et al found that the majority of children with multiple islet cell autoantibodies developed T1 DM within 15 years.11 Of note, a small percentage of patients exhibit destruction of the beta cells without autoantibodies, which is referred to as T1B DM. These patients still have an insulin deficiency, but the pathogenesis is idiopathic and occurs mainly in Asian or African people.2,9 Because the destruction of the beta cells is the cornerstone of T1 DM, it is not a reversible condition like T2 DM.
Like T1 DM, the etiology of T2 DM also is multifactorial. There is a strong hereditary component, and newly diagnosed youth often have an identified first- or second-degree relative with diabetes, with a frequency of 74% to 100%.12 Hormonal changes that occur during puberty also are contributory. Hyperinsulinemia develops a secondary increased insulin resistance. This insulin resistance has been attributed to the increased secretion of growth hormone during puberty.13 Also, as previously highlighted, insulin sensitivity varies by race. African American children, from 7 to 11 years of age, have been found to have higher insulin levels than age-matched white children.13 There is a high correlation between obesity and the development of T2 DM. Children who are obese, particularly those with high weight, height, and waist circumference, are more prone to have insulin resistance.14 Other environmental factors, including sedentary lifestyles that lead to obesity, have been associated with living in low-income neighborhoods. Children who attend schools in higher socioeconomic status (SES) environments have more regular physical education classes than children who attend schools in lower SES neighborhoods. These same lower SES neighborhoods often have limited access to grocery stores that provide healthy, nutritional food offerings.15
Associated Illnesses
Because T1 DM is an autoimmune condition, it has been associated with other autoimmune diseases as well. Autoimmune thyroid disease and celiac disease are the most frequently associated autoimmune disorders in patients with T1 DM. Hypothyroidism occurs in about 12% to 24% of females and 6% of males, whereas hyperthyroidism has a prevalence of 1% to 2% in all T1 DM patients.16 In T1 DM, patients with GAD and ZnT8 autoantibodies were found to have an increased risk for autoimmune thyroiditis.9 Celiac disease has a prevalence of 8% in the T1 DM population, and patients often are screened for it when initially diagnosed.17 Autoimmune polyendocrine syndromes (APS) are a more rare affiliation with T1 DM. APS represents a heterogenous group of diseases characterized by an insufficiency of multiple endocrine organs due to an immunologic destructive process. T1 DM is one of the diseases associated with the different types of APS, particularly type 2 APS.9,18 It is not uncommon when a patient is diagnosed with DM to have regular screening for one of the aforementioned illnesses.
Metabolic syndrome is a set of disease risk factors that, when present, is highly predictive of developing T2 DM.19,20 The etiology of metabolic syndrome is insulin resistance or hyperinsulinemia. The American Heart Association characterizes metabolic syndrome as including central or abdominal obesity, dyslipidemia, hypertension, and glucose intolerance. Not only does metabolic syndrome increase the risk of developing T2 DM, the components also contribute to the morbidity of patients during disease progression.
Morbidity
When left untreated or poorly controlled, diabetes — regardless of type — can lead to many complications, such as hypertension, dyslipidemia, retinopathy, nephropathy, neuropathy, and poor wound healing.4 Many of these complications arise later in life in T1 DM, but can manifest sooner with poor glycemic control.
Children with T1 DM are not only at increased risk for complications directly related to glucose metabolism and other autoimmune diseases as discussed earlier, but they also are found to have an increased prevalence of mental health disorders, such as anxiety, depression, and eating disorders.21,22 Unfortunately, these mental disorders have demonstrated a negative impact on glycemic control.22,23 Patients with T2 DM also have increased rates of depression and anxiety compared to the general population.24 Hence, this stresses the importance of screening for mental health comorbidities in addition to the complications that arise from glucose metabolism.
There are similar comorbidities associated with T2 DM, for which youth should be evaluated. Ten percent to 15% of youth have albuminuria at the time of diagnosis. The prevalence increases over the course of the illness, leading to the development of nephropathies. Twenty percent to 30% of youth have hypertension at the time of diagnosis, which, if left untreated, accounts for a great proportion of micro- and macrovascular complications. Twenty percent to 45% of youth with T2 DM have hepatic steatosis, which can lead to the development of nonalcoholic fatty liver disease and cirrhosis.25 A national sample of American youth recently diagnosed with T2 DM found that 80% had low high-density lipoprotein cholesterol and 10% had high triglycerides.26 This underscores the importance of screening for dyslipidemia.
Mortality
Children with DM are more likely to die from hypoglycemia and DKA, which are largely preventable. In a Centers for Disease Control and Prevention (CDC) analysis of deaths caused by DM in youth aged 1 to 19 years, the death rate was approximately one per 1 million population during 2012 to 2014. This has decreased when compared to previous years. However, there were significant racial/ethnic disparities; death rates among blacks were approximately twice as high as those of whites and hispanics.27 The CDC did not distinguish this mortality rate between T1 DM and T2 DM, but there is a higher incidence of T1 DM than T2 DM in this age group.
Pathophysiology
As previously mentioned, T1 DM results from the destruction of beta cells of the endocrine pancreas, whereas T2 DM is characterized by insulin resistance. Regardless of etiology, insulin deficiency or resistance leads to an inability to use glucose. The body’s inability to metabolize glucose causes an increase in counter-regulatory hormones, such as glucagon, catecholamines, growth hormone, and cortisol, as depicted in Figure 2.28 These counter-regulatory hormones lead to an increased catabolic state leading to increased glucose production by the liver and the kidneys, resulting in marked hyperglycemia and hyperosmolality.29 This hyperglycemia causes extracellular fluid and electrolyte shifts. Despite this surplus of glucose, the lack of insulin means the body cannot use the glucose, leading to “internal starvation.”30
Figure 2. Pathophysiology of Diabetic Ketoacidosis |
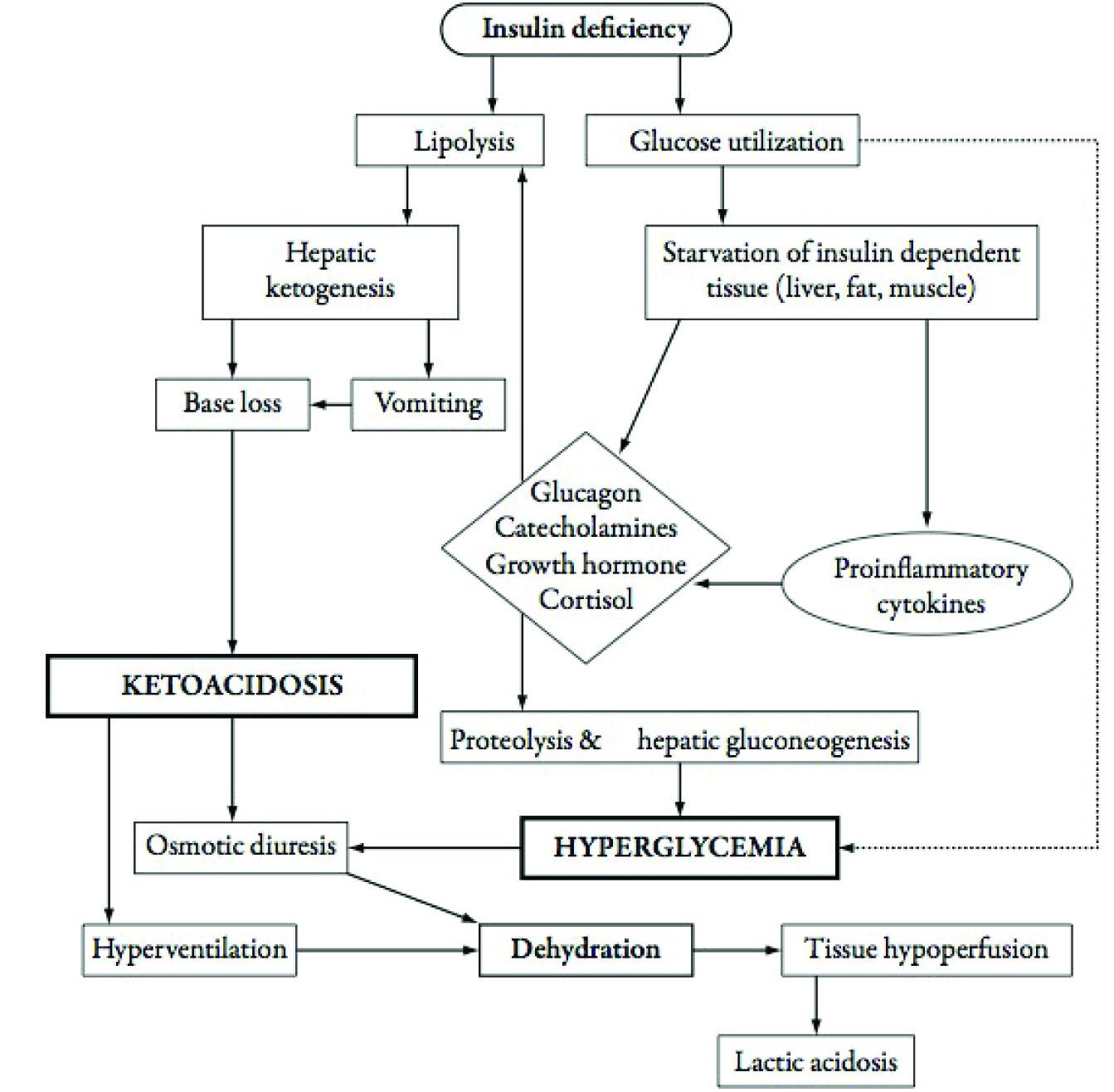 |
|
Reprinted from Rosenbloom A. The management of diabetic ketoacidosis in children. Diabetes Ther 2010;1:103-120. |
These counter-regulatory hormones also induce lipolysis leading to an increase in free fatty acids, which can be used as an energy source for many tissues except for the brain. Without insulin, the increase in lipolysis diverts elevated free fatty acids to ketogenesis in the liver. The primary ketones produced are acetoacetate and β-hydroxybutyrate.29-33 Spontaneous breakdown of acetoacetate to acetate in the lungs is responsible for the pathognomonic clinical presentation of fruity breath in DKA. The brain uses these ketone bodies as a substrate for energy since it cannot use free fatty acids. However, the ketone bodies are acidic molecules, and because of significant overproduction, the body is unable to buffer these weak acids, leading to ketoacidosis. Normally at physiologic pH, these ketone bodies dissociate.31 Additionally, the lack of insulin leads to decreased clearance of ketone bodies, but the mechanism is not known.32 The ratio of β-hydroxybutyrate to acetoacetate is typically 1:1 in physiologic ketosis that occurs during times of fasting and prolonged exercise, but it can be as high as 10:1 in DKA.31
Elevated ketoacidosis also causes osmotic diuresis and dehydration. The dehydration worsens, causing poor tissue perfusion, leading to lactic acidosis and renal dysfunction from prerenal acute kidney injury, which further contributes to the patient’s acidosis. This metabolic acidosis induces respiratory compensation. Initially, the patient has rapid, shallow respirations, but in severe metabolic acidosis, this can progress to deep and labored respirations called Kussmaul respirations. Severe metabolic acidosis or DKA can lead to coma and death if not treated.
There are instances in which people intentionally want their body in ketosis. The ketogenic diet commonly is used to control epilepsy refractory to antiepileptic medications and, more recently, for weight loss. This differs from DKA because the degree of ketosis in these instances does not overwhelm the body, and the ketones still can be eliminated. The mechanism of how the ketosis becomes neurotoxic is not clear.30
In T2 DM, patients are less likely to progress to DKA due to a relative insulin deficiency from insulin resistance vs. a complete deficiency as seen in T1 DM. As seen in Figure 3, genetics and lifestyle (increased sedentary behavior, excess energy intake) play a key role in the pathogenesis of T2 DM.33 This peripheral resistance leads to the pancreas producing increased insulin secretion. This sustained hyperinsulinemia can result in β-cell exhaustion and decreased ability to secrete insulin, ultimately leading to T2 DM.3
Figure 3. Pathogenesis of Type 2 Diabetes Characterized by Impaired Insulin Secretion and Insulin Resistance |
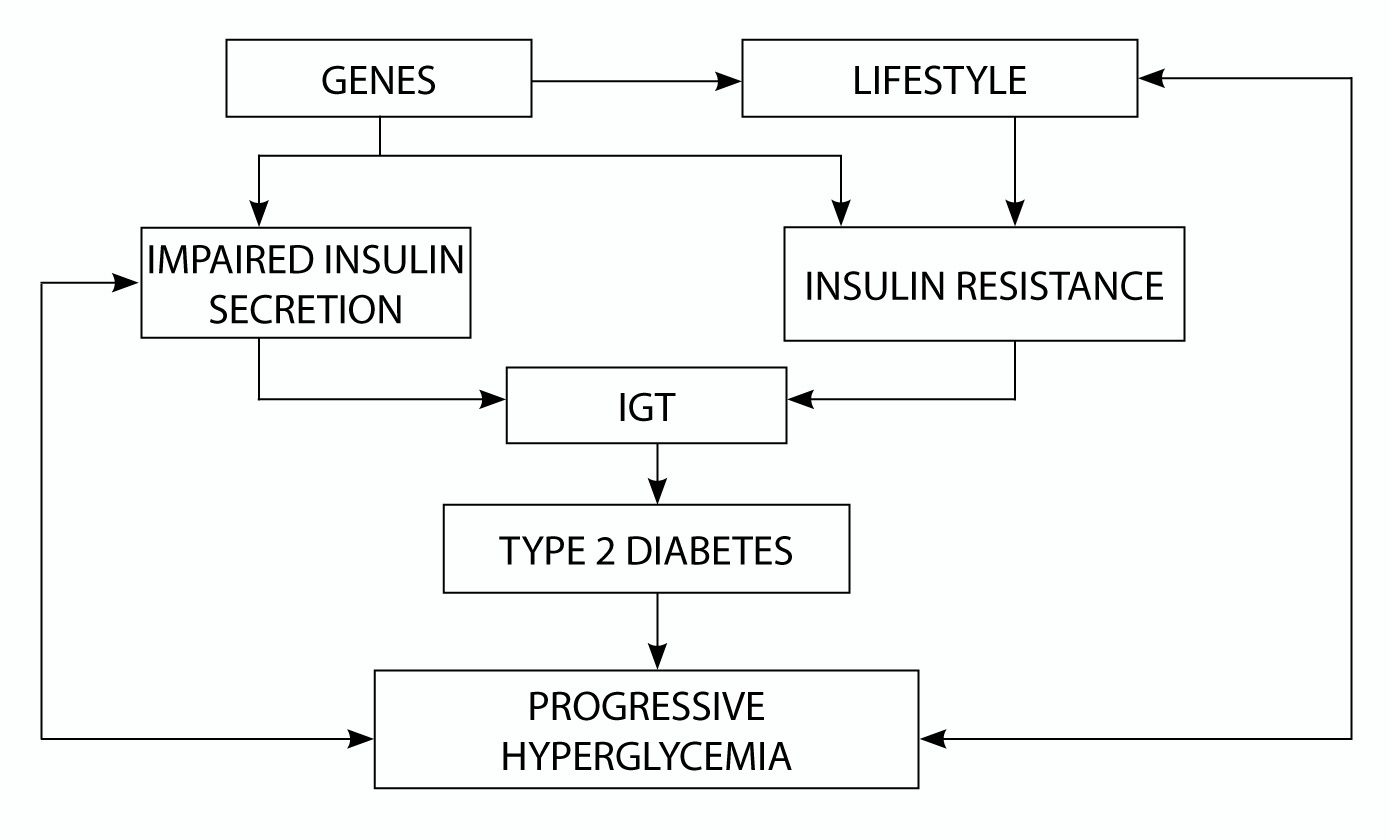 |
|
IGT: impaired glucose tolerance |
Special Considerations
T1 and T2 DM are the most common types of DM seen in the pediatric population. However, there are other types of DM in children and adolescents, such as monogenic diabetes and drug-induced diabetes.
Monogenic Diabetes
T1 and T2 DM are both polygenic. However, there is another entity of diabetes called monogenic diabetes, which includes neonatal diabetes and a familial form of nonketotic DM formerly called maturity onset diabetes of the young (MODY).3 The estimated monogenic DM population prevalence is about 1.2%.1 Monogenic diabetes refers to the inheritance of a mutation or mutations in a single gene confirmed by molecular testing.
Monogenic diabetes is now classified by its genetic subgroups. Hepatic nuclear factor (HNF)-1α MODY and HNF-4α MODY contribute to the majority of cases. The classic phenotype includes onset before age 25 years; nonketotic, noninsulin-dependent diabetes; and an affected parent.34 However, not all cases fit this classic phenotype. In the past, patients would be misdiagnosed with T1 and T2 DM. Suspicion should be raised particularly with patients with three or more generations affected.34 The importance in diagnosing monogenic diabetes is that certain genetic forms are managed differently and have a different clinical course. For instance, children with HNF-1α MODY and HNF-4α MODY respond better to sulfonylureas than insulin.3,35
Neonatal diabetes occurs within the first three months of life and its prevalence is 0.2%.1,3,34,35 There are two main types: transient and permanent. Transient resolves by 12 weeks of age but has a 50% relapse rate. Genetic testing should be done for all infants diagnosed with DM, particularly those younger than 6 months of age. The most common causes of neonatal DM respond to sulfonylureas, and early treatment may improve the associated neurocognitive defects.36
Drug-Induced Diabetes
Numerous medications can cause diabetes; however, the mechanisms by which this occurs vary. Medications such as glucocorticoids and atypical antipsychotics can result in insulin resistance. Beta blockers decrease insulin secretion, and chemotherapeutic agents, such as tacrolimus and cyclosporine, can directly damage islet cells. Patients who require any such medications should be screened cautiously.37,38
Clinical Features
Case 1
A 13-year-old girl presents to the emergency department (ED) with complaints of thick, white vaginal discharge and itching. She presented with similar symptoms two months ago and was diagnosed with vaginal candidiasis. During the exam, the mother mentions her concern that the patient has been more fatigued. She reports about a 20-pound unintentional weight loss over the past few months. The mother is concerned about this weight loss because the patient is “already so skinny.” The patient has been eating more despite the weight loss. Her mother also has noticed that the patient has been using the restroom more frequently and always appears to be thirsty. On exam, the patient is awake and alert with normal mentation. Her mucous membranes are dry, and her eyes appear slightly sunken. On genital exam, patient has normal external female genitalia and thick, white discharge is seen in the vaginal vault. The remainder of her exam is unremarkable. Her vital signs show a heart rate of 120 beats per minute, respiratory rate of 18 breaths per minute, and blood pressure of 114/82 mmHg. Her urinalysis shows large ketones and 3+ glucose.
Type 1 Diabetes Mellitus
There are multiple red flag symptoms for new-onset T1 DM in this case. Patients often present with a constellation of symptoms, including weight loss, polydipsia, polyuria, polyphagia, fatigue, and dehydration. Glucosuria occurs when the kidneys exceed their capacity to absorb glucose. Since glucose acts an osmotic diuretic, the glucosuria is accompanied by water loss. This water loss activates thirst and leads to polydipsia. The negative calorie balance from the glucosuria and tissue catabolism leads to polyphagia.33 Hyperglycemia from the uncontrolled DM can make patients susceptible to infections. In case 1, the patient had recurrent vaginal candidiasis. Consider the possibility of DM in recurrent cases and also in prepubertal girls with vaginal candidiasis.3 Additionally, the patient has ketonuria, which is associated more commonly with T1 DM, as seen in Table 1.
Table 1. Clinical Characteristics of Type 1 Diabetes, Type 2 Diabetes, and Monogenic Diabetes in Children and Adolescents |
|||
|
Characteristic |
Type 1 |
Type 2 |
Monogenic |
|
Genetics |
Polygenic |
Polygenic |
Monogenic |
|
Age of onset |
6 months to young adulthood |
Usually pubertal (or later) |
Often post pubertal except glucokinase and neonatal diabetes |
|
Clinical presentation |
Most often acute, rapid |
Variable; from slow (often insidious) to severe |
Variable (may be incidental in glucokinase) |
|
Autoimmunity |
Yes |
No |
No |
|
Ketosis |
Common |
Uncommon |
Common in neonatal diabetes, rare in other forms |
|
Glycemia |
High |
Variable |
Variable |
|
Obesity |
Population frequency |
Increased frequency |
Population frequency |
|
Acanthosis nigricans |
No |
Yes |
No |
|
Frequency (% of all diabetes in young people) |
Usually 90%+ |
Most countries < 10% (Japan 60-80%) |
1-2% |
|
Parent with diabetes |
2-4% |
80% |
90% |
|
Adapted from International Diabetes Federation/International Society for Pediatric and Adolescent Diabetes. Global IDF/ISPAD Guideline for Diabetes in Childhood and Adolescence. 2011. p12. Available at: https://cdn.ymaws.com/www.ispad.org/resource/resmgr/Docs/idf-ispad_guidelines_2011_0.pdf. |
|||
Case 2
A 6-year-old boy is emergently roomed in the resuscitation bay by the triage nurse because of altered mental status. His mother reports that he was diagnosed with influenza two days ago at another ED and was prescribed Tamiflu. The patient has been complaining of abdominal pain and vomiting. His mother reports that the patient has been acting more lethargic, and today she noticed that he was breathing oddly and decided to bring him in for further evaluation. On exam, the patient is intermittently opening his eyes but is not alert. He occasionally moans and at times will follow commands. He has deep and labored breathing and his breath smells sweet and fruity. His vital signs show a heart rate of 150 beats per minute, respiratory rate of 48 breaths per minute, and blood pressure of 101/72 mmHg. His blood gas shows pH 7.01, pCO2 25, base deficit 14, and glucose 642.
Diabetic Ketoacidosis
This patient is presenting in severe DKA. He has a waxing and waning mental status, Kussmaul respirations, fruity breath, tachycardia, and impressive acidosis. Younger children tend to present in severe DKA due to acute onset of severe insulin deficiency and the diagnosis not being considered in the early stages. Another pitfall to avoid is attributing the Kussmaul respirations to an asthma exacerbation or pneumonia. With Kussmaul respirations, patients will have clear breath sounds and often will not have concomitant upper respiratory infections. The severe metabolic acidosis leads to respiratory compensation with initially rapid, shallow breathing that progresses to the deep and labored breathing consistent with Kussmaul respirations. Vomiting may be misattributed to the beginnings of acute gastroenteritis. Consider the diagnosis of DM in the differential for patients presenting with vomiting and dehydration, especially without the presence of diarrhea. The abdominal pain associated with DKA can simulate an acute abdomen.35 This abdominal pain should resolve as the acidosis improves. If it does not, consider another etiology for the abdominal pain. The waxing and waning mental status is concerning for the development of another complication called cerebral edema. Cerebral edema will be discussed in further detail later in this article.
Case 3
A 12-year-old female is brought to the ED by her grandparent with a chief complaint of a rash. The patient reports the rash is not pruritic, is not associated with the introduction of new foods or dyes, and it has persisted over the last year despite her use of over-the-counter hydrocortisone. On physical exam, the patient has a rash on the nape of her neck that is hyperpigmented and has a velvety appearance. She is notably hirsute and appears overweight, but the remainder of her exam is otherwise unremarkable. Her vital signs show heart rate of 96 beats per minute, respiratory rate of 18 breaths per minute, and blood pressure of 138/96 mmHg. Her fingerstick glucose is 227 and her urinalysis reveals glucosuria.
Type 2 Diabetes Mellitus
T2 DM in childhood most often presents in the peripubertal age. At the time of diagnosis, most youth are obese, and have components of metabolic syndrome as evidenced in this vignette by hypertension. The symptoms of polyuria and polydipsia often are absent or very mild. Screening urinalysis typically reveals glucosuria without ketonuria. Contrary to T1 DM, there is usually no history of significant weight loss. Other characteristics commonly seen in patients with T2 DM are acanthosis nigricans and polycystic ovarian syndrome (PCOS). Acanthosis nigricans is a skin disease found on the lateral and posterior neck, axilla, and groin. It is most common in obese patients with insulin resistance, and up to 90% of children with T2 DM develop it over the course of their illness. PCOS also is associated with insulin resistance and obesity. PCOS is a disorder of hyperandrogenism and chronic anovulation, and can result in menstrual cycle changes, increased facial and body hair, acne, cysts in the ovaries, and infertility.
Diagnosis
When patients present without distinct symptoms of severe hyperglycemia, the diagnosis of DM can be made based on the presence of one of the four methods, and must be confirmed by repeat testing on a different day:12,39
- fasting glucose level ≥ 126 mg/dL;
- two-hour post challenge glucose ≥ 200 mg/dL;
- hemoglobin A1c (HbA1c) > 6.5%;
- minor symptoms (mild polyuria and polydipsia) and a random serum glucose ≥ 200 mg/dL.
Once DM is determined, clinical features and further testing can help determine the exact type. Table 2 shows how to further differentiate the different types of DM. The presence of autoantibodies can help distinguish between T1 DM and T2 DM. Islet cell autoimmunity occurs with relative frequency in youth who phenotypically appear to have T2 DM; testing should be considered in these patients, since their presence indicates an earlier need for insulin. T1 DM can be confirmed with the presence of at least two autoantibodies.40 Studies suggest that initial evaluation should start with testing for GAD and IA2 autoantibodies initially, since these often are seen in the initial diagnosis of T1 DM. Within pancreatic beta cells, proinsulin is split apart to form one molecule of C-peptide and one molecule of insulin. When insulin is released from the beta cells into the blood, equal amounts of C-peptide also are released. Since C-peptide is produced at the same rate as insulin, it is useful as a marker of insulin production. C-peptide levels are elevated in patients with T2 DM and when present, can help delineate the type of diabetes and direct further treatment.
Table 2. Classification of Pediatric Diabetes |
|
|
Type 1 Diabetes |
Antibodies present (GADA, IA2A, ZnT8, IAA) |
|
Type 2 Diabetes |
No antibodies Insulin resistant (large waist) |
|
MODY |
No antibodies Insulin sensitive (normal waist) Additional testing |
|
Secondary Diabetes |
No antibodies Insulin sensitive (normal waist) Additional testing |
|
MODY: maturity onset diabetes of the young; GADA: glutamic acid decarboxylase antibody; IA2A: insulinoma-associated-2 antibodies; ZnT8: zinc transporter-8; IAA: insulin autoantibody |
|
Diabetic Ketoacidosis
DKA is the most common complication of T1 DM; it occurs in about 25% to 40% of children.41 Untreated T1 DM progresses to DKA. It occurs predominantly in patients with T1 DM but has been known to occur in patients with T2 DM as well. It is the most common cause of hospitalization and death in patients with diabetes.42 As detailed in the pathogenesis section, if the body is unable to use glucose, the resulting counterregulatory hormones stimulate further production of glucose and increase the production of ketone bodies. The production of the ketone bodies overwhelms the body, causing an anion-gap metabolic acidosis or DKA.
The diagnosis of DKA is confirmed by the following:43,44
- pH < 7.3 or bicarbonate < 15 mmol/L;
- glucose > 200 mg/dL (> 11 mmol/L);
- ketonemia (β-hydroxybutyrate > 3 mmol/L) and ketonuria (> 2+, moderate or large).
The severity of DKA is categorized by the severity of the acidosis:43,44
- Mild: pH < 7.3 or bicarbonate < 15 mmol/L;
- Moderate: pH < 7.2 or bicarbonate < 10 mmol/L;
- Severe: pH < 7.1 or bicarbonate < 5 mmol/L.
DKA is more likely to be present in the initial onset of T1 DM in younger children, children without a first-degree relative with T1 DM, children in a lower SES, and children in countries with a low prevalence of T1 DM.43,44 Patients not taking their insulin, insulin pump failure, and failure to follow sick day management account for nearly all cases of recurrent DKA.43
Patients in DKA have an anion-gap acidosis due to the production of ketone bodies. Calculation of the anion gap is: Anion gap = Na – (Cl + HCO3). The normal range is 12 +/- 2 mmol/L. In DKA, the bicarbonate is replaced by β-hydroxybutyrate, causing the increased anion gap. In addition to an anion gap metabolic acidosis, there are other laboratory abnormalities seen on the basic metabolic panel (BMP). The sodium will be falsely lowered because of the hyperglycemia. To calculate the true sodium level, use the following formula: corrected Na = measured Na + 2 [(plasma glucose – 100) / 100] mg/dL. The patient’s sodium should correct as the patient’s acidosis is corrected. Phosphate, magnesium, and calcium are other elements excreted in excess in urine during the development of DKA.45 Additionally, if a complete blood cell count (CBC) is ordered, it not unusual that the white blood cell (WBC) count will be elevated in DKA because of a stress response.43
Hyperglycemic Hyperosmolar State
Hyperglycemic hyperosmolar state (HHS) is a potentially fatal hyperglycemic crisis that occurs as an acute complication of uncontrolled DM. Insulin deficiency and subsequent hyperglycemia leads to glycosuria, osmotic diuresis, and dehydration in both DKA and HHS.45 However, the clinical presentation of HHS differs from DKA. HHS tends to occur in obese patients and often is the first presentation of previously undiagnosed T2 DM. There is a subtle, gradual, and often unnoticed increase in polyuria and polydipsia, ultimately associated with significant dehydration. The fluid loss in patients with HHS is estimated to be twice that of patients with DKA.46 The diagnostic criteria for HHS include plasma glucose concentration > 600 mg/dL, serum carbon dioxide concentration > 15 mmol/L, small ketonuria, absent to low ketonemia, effective serum osmolality > 320 mOsm/kg, and an alteration in mental status resulting in combativeness, seizures, obtundation, or coma.43,47 The most common precipitating event is an underlying infectious illness that causes a higher metabolic stress/demand leading to profound dehydration.48 However, there are other potential triggers for HHS, which include silent myocardial infarction, cerebrovascular accident, mesenteric ischemia, acute pancreatitis, and use of certain medications, such as steroids and calcium channel blockers.45 While the incidence rate of HHS is less than DKA, the mortality rate is much higher. The associated acidosis in DKA usually leads to a much earlier diagnosis; the insidious onset of HHS likely contributes to its more severe course.
Management
T1 DM is treated by providing exogenous insulin. Patients must monitor their blood glucose measurements closely and take insulin accordingly. The first steps in the treatment of T2 DM are lifestyle and behavior modification, targeting increased physical activity and healthy eating habits to promote optimal weight management. Pharmacologic intervention often is required, and the first-line agent is metformin. The treatment goal in the pediatric population is to achieve a target hemoglobin A1c level lower than 7.5% per the American Diabetes Association.40,49 DKA and HHS are conditions that are encountered frequently in the pediatric ED that require prompt identification and management to prevent further complications. (See Figure 4.)
Figure 4. Management of Type 2 Diabetes Mellitus by Blood Glucose |
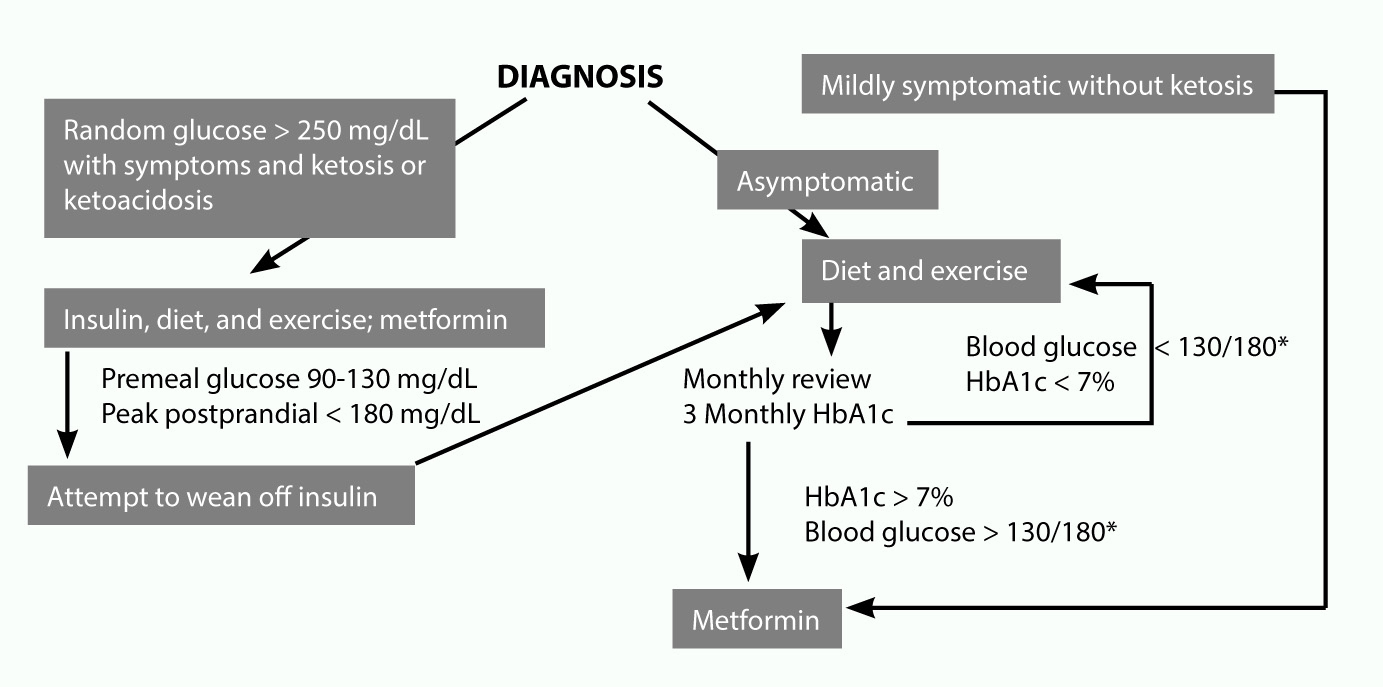 |
|
*Blood glucose values < or > 130/180 refer to self-monitoring of plasma blood glucose values of 90 mg/dL to 130 mg/dL fasting or preprandial and peak postprandial values of < 180 mg/dL. |
Diabetic Ketoacidosis
In severe cases of DKA, it is important to identify first if the patient can maintain his or her airway. Patients in DKA often are very tachypneic to maintain a respiratory alkalosis to compensate for their metabolic acidosis. Do not rush to intubate a patient in DKA. It can be difficult to maintain this same degree of respiratory alkalosis with mechanical ventilation. Additionally, there is concern that intubation could lead to a sudden increase in pCO2, causing cerebral spinal fluid pH to decrease and contribute to worsening cerebral edema.43 However, if the patient appears to be tiring out and unable to maintain this compensation, this is an indication to intubate the patient. Otherwise, avoid endotracheal intubation and consider noninvasive ventilation to support the patient.
Establish parenteral access, intravenous (IV) or intraosseous (IO), as soon as possible. Multiple access points are preferred because of the need for a continuous insulin infusion and IV fluids, which is discussed later in this article. Avoid placing central lines because of the increased risk for thrombosis.43 In addition to obtaining parenteral access, order the following laboratory tests: blood gas, BMP, urinalysis (UA), and HbA1c. Consider a CBC, blood culture, or urine culture if there are any concerns for an infection, and a pregnancy test in pubertal females. While obtaining access, it is important to obtain a full history from the caregiver to identify a potential underlying cause and perform a complete physical exam, particularly noting the patient’s mental status and hydration status. Calculating the patient’s Glasgow Coma Scale (GCS) score is recommended as an objective measure of the patient’s mental status. Vital signs, neurologic status, and blood glucose levels all need to be monitored hourly.
DKA treatment goals include rehydration, inhibition of the counter-regulatory hormones that increase the production of ketone bodies, reversal of electrolyte abnormalities, and, ultimately, correction of the anion gap metabolic acidosis.29,31 The reversal of the aforementioned treatment goals requires IV fluids and insulin. There are special considerations regarding these treatments in the pediatric population, as follows.
Fluids. The administration of fluids is vital in the initial resuscitation of patients in DKA. Fluid replacement should commence prior to starting insulin. Common regimens advise giving an initial 10 mL/kg to 20 mL/kg isotonic fluid bolus over one to two hours.50 Optimal volume and fluid administration rate are somewhat controversial and variable. One common option is to administer one to two times maintenance fluids after the initial bolus. Another option is to estimate the degree of dehydration and use this to calculate the fluid deficit. This fluid deficit, in addition to maintenance fluid requirements, is replaced over the next 24 to 48 hours.31,50
Once the patient’s blood glucose falls below 250 mg/dL, dextrose-containing IV fluids will need to be added to prevent hypoglycemia. The two-bag system involves two bags of identical fluids with electrolytes except that one bag contains 0% dextrose and the other bag contains 10% dextrose. This allows customization of the dextrose concentration to meet the patient’s needs. Typically, once the blood glucose level is below 250, the total fluid rate is split equally across both bags, essentially making a 5% dextrose fluid. If the blood glucose continues to drop more than 100 mg/dL per hour, then the full calculated rate will be provided by the 10% dextrose bag only.31
Recommendations for fluid management are based on retrospective data. The Pediatric Emergency Applied Research Network (PECARN) Fluid Therapies Under Investigation in DKA (FLUID) study is the first prospective, randomized controlled clinical trial investigating the impact of different fluid rehydration regimens on the neurological and cognitive outcomes of children in DKA.50-52 This study analyzed the effect of not only fluid infusion rate, but also the sodium content of fluids.51 All children received an initial 10 mL/kg bolus of 0.9% saline before being randomized to one of four treatment protocols. Ultimately, the study found that neither the infusion rate nor the sodium chloride content had an effect on the neurologic outcomes of children in DKA.53
Insulin. Insulin therapy should begin after fluid therapy is initiated. The insulin drip should be started at 0.1 units/kg/hour. A bolus dose of insulin is not recommended. If the patient’s blood glucose drops below 250 mg/dL or if the glucose is decreasing by more than 100 mg/dL, do not decrease the insulin drip. Instead, start the dextrose-containing IV fluids as part of the two-bag method described previously. The insulin will inhibit the counter-regulatory hormones and halt the production of ketone bodies and restore normal cellular metabolism. Continue the insulin drip until there is resolution of DKA evidenced by the following: pH > 7.3, bicarbonate > 15 mmol/L, or closure of the anion gap. The resolution of this acidosis often takes longer than the normalization of the patient’s glucose.44
Electrolyte Management. Sodium, potassium, chloride, phosphorus, calcium, and magnesium are affected in DKA. Potassium may seem elevated despite a total body deficit.48 Potassium losses are through vomiting and osmotic diuresis. It important that potassium is added to the IV fluids to prevent hypokalemia. Potassium can be provided as a combination of potassium chloride, potassium phosphate, or potassium acetate at 40 mEq/L to 80 mEq/L.28,54 If the patient is hyperkalemic, do not add potassium to IV fluids until the patient urinates. Hypokalemia can develop with initiation of insulin. It is important to monitor potassium levels to prevent this. Phosphate and magnesium do not need replacement routinely in DKA. Consider phosphate repletion if the patient has unexplained weakness or decreased cardiac function.31
Hyperglycemic Hyperosmolar State. As noted earlier, the first step in managing any hyperglycemic emergency is to secure the patient’s airway and ensure adequate oxygenation and ventilation. While securing IV access, serum electrolytes and arterial/venous blood gas, urinalysis, and HbA1c should be obtained. Additional laboratory tests, such as a CBC and blood and urine cultures, should be pursued if there is a suspected infection. It is important to note that leukocytosis can result from either the underlying infection or elevated stress hormones.
The primary goals in the management of HHS are to ensure adequate fluid resuscitation, administer insulin, correct electrolyte derangements, and identify and treat the underlying cause. The indicated initial fluid management is a 20 mL/kg bolus of isotonic saline (0.9% NaCL), which may be repeated until the patient’s perfusion and hemodynamic status have been restored. Once this occurs, the remaining fluid losses can be replaced with 0.45% NaCL over the course of 24 to 48 hours, while frequently measuring the serum sodium levels to ensure a gradual decline in the accompanying hypernatremia. It also is recommended that urinary losses be replaced with 0.45% NaCL.43,46 This differs from the recommended management of DKA. While there is a whole-body depletion of plasma potassium, in hyperglycemic emergencies patients often will present with hyperkalemia that resolves quickly with fluid resuscitation and insulin administration. The serum glucose usually is higher in HHS than it is in DKA and declines notably with fluid resuscitation. Once the serum glucose is no longer decreasing at a rate of at least 50 mg/dL per hour with fluids alone, a continuous infusion of insulin at 0.025 to 0.05 U/kg/hour should be administered.43 Patients with HHS have more significant deficits of potassium and phosphate than patients with DKA.43 Once the serum potassium level has fallen to a normal range, start replacement therapy. This can be done most effectively by administering a 50:50 mixture of either potassium chloride or potassium acetate and potassium phosphate. By instituting phosphate replacement, one can avoid the potential complications of hypophosphatemia, which include muscle weakness, rhabdomyolysis, and hemolytic uremia.
Complications
Cerebral Edema
Cerebral edema is the most feared complication of T1 DM. Cerebral edema has varying severity and can be present subclinically.50,55 Symptomatic cerebral edema is considered the manifestation of the severe form that includes the following: waxing and waning mental status, decorticate and decerebrate posturing, abnormal motor or verbal response to pain, and abnormal neurologic respiratory patterns.50,56 Typically, clinical cerebral edema occurs four to 12 hours after initial presentation, but it can be present prior to the initiation of therapy or as late as 24 to 48 hours afterward.43,52 Symptomatic cerebral edema is a rare complication of DKA with an incidence of approximately 1%, yet the mortality is disproportionately higher at 21% to 24%.41,43,57
The pathogenesis of cerebral edema is not known. Generally, it was thought to be a consequence of inappropriate DKA treatment due to rapid osmotic changes and fluid shifts sustained when patients were given a large volume of IV fluids. However, the presence of cerebral edema prior to the start of therapy and despite the initiation of fluid protocols suggests an alternative mechanism.58,59 It has been hypothesized that vasogenic edema from destruction of the blood brain barrier also could lead to cerebral edema. The abnormal diffusion of fluid into the brain seen on magnetic resonance imaging supports this, but these studies were small.50 Recent studies suggest that the development of cerebral edema may be related to the initial severity of dehydration and acidosis, causing cerebral hypoperfusion and triggering injury and edema, similar to cytotoxic edema in ischemic stroke.50,59 It is likely that the development of cerebral edema is multifactorial.
There is an increased association with cerebral edema in patients presenting with severe acidosis, severe hypocapnia, higher blood urea nitrogen, lower initial bicarbonate, a new diagnosis of diabetes, and a slow increase in serum sodium concentration with treatment.50,57 Many of these factors are related to dehydration leading to decreased cerebral perfusion. Young age and treatment with bicarbonate also are risk factors for cerebral edema. Because of this, bicarbonate is not recommended in the treatment of DKA. It is theorized that young children’s brains may be more susceptible to the metabolic and vascular changes in DKA, increasing their risk for cerebral edema.50 Additionally, symptomatic cerebral edema rarely occurs in patients older than 20 years of age.56
Since the pathogenesis is not well understood, focus on the quick identification and treatment of clinical cerebral edema to reduce morbidity and mortality. Once identified, patients should receive hyperosmolar therapy with either hypertonic saline (3% NaCl) 5 mL/kg to 10 mL/kg or mannitol 1 g/kg over 10 minutes. Head imaging only should be obtained once the patient is stabilized and hyperosmolar therapy has been initiated. The imaging should be obtained to rule out other etiologies for the patient’s altered mental status.41,43 Conversely, about half of patients in DKA with or without neurological symptoms can show narrowing of the lateral ventricles on head imaging hours after initiation of therapy substantiating that subclinical cerebral edema occurs.41,60 If a patient is showing clinical signs of cerebral edema, do not delay hyperosmolar treatment for head imaging.
Hypoglycemia
The International Hypoglycemia Study Group (IHSG) suggested three levels of hypoglycemia to address the issue of hypoglycemic risk. Clinical hypoglycemia alert is defined as a blood glucose less than 70 mg/dL. Clinically important hypoglycemia is defined as blood glucose less than 54 mg/dL. Severe hypoglycemia is defined as an event associated with severe cognitive impairment requiring the assistance of another person to take corrective actions.61 If left untreated, hypoglycemia can lead to death. The treatment of hypoglycemia depends on the patient’s mental status. If the patient is awake and conscious, have the patient ingest a concentrated and rapidly absorbed simple carbohydrate, such as juice. However, if the patient is unconscious, administer glucagon, or if the patient has IV access, give a dextrose bolus. It is imperative that a blood glucose level is repeated within 15 to 20 minutes to ensure the hypoglycemia has been corrected.
Hyperglycemic Hyperosmolar State
There are unique complications associated with HHS, such as rhabdomyolysis, which is encountered commonly. It is recommended that screening creatine kinase concentrations be monitored every two to three hours. There also have been documented cases of children with HHS who develop malignant hyperthermia. These patients can be treated with dantrolene; however, there is a high mortality rate despite treatment.46 Patients with central venous catheters are at an increased risk for developing venous thrombosis similar to in DKA. Heparin administration should be considered in patients with HHS who require central venous catheters and are immobile for more than 24 to 48 hours.
Disposition
Patients who present with hyperglycemic emergencies, severe dehydration, and metabolic derangement should be admitted to the hospital in an intensive care unit (ICU) setting. The American Diabetes Association admission guidelines for DKA are a plasma glucose concentration greater than 250 mg/dL, a pH level below 7.30, a serum bicarbonate level of less than 15 mEq/L, and a moderate or greater level of ketones in the serum or urine.62 Resolution of DKA is defined as a blood glucose concentration of less than 200 mg/dL, a bicarbonate level of 18 mEq/L or greater, and a venous pH level of greater than 7.3. Patients typically can be discharged to home once the DKA has resolved, they can tolerate oral intake, and they have been transitioned to subcutaneous insulin. Children with newly diagnosed DM who do not present in extremis do not need ICU monitoring. However, admission for multidisciplinary studies (endocrinology, nutrition) should be strongly considered.
Summary
DM is one of the most common chronic diseases in children and adolescents. The primary two forms of DM encountered in the ED are T1 DM and T2 DM. The majority of children will have T1 DM; however, the incidence of T2 DM is increasing. T1 DM is an autoimmune condition and is associated with other autoimmune conditions, such as thyroiditis and celiac disease. Contrarily, T2 DM is caused by insulin resistance and a failure of pancreatic compensatory mechanisms.
In both disease conditions, the body cannot effectively metabolize glucose. In the case of T1 DM, the pancreas no longer produces insulin due to autoimmune destruction of the beta cells of the endocrine pancreas. Patients with T1 DM require insulin as the mainstay of their treatment. However, insulin resistance is the primary dysfunction in T2 DM. Often, patients take oral medications such as metformin to treat their disease with severe cases, ultimately needing insulin similar to patients with T1 DM.
Patients can have life-threatening complications in both conditions. DKA is associated mainly with T1 DM; however, a small percentage of patients with T2 DM can present in DKA. Providers should treat patients in DKA with IV fluids for rehydration, a continuous infusion of insulin, and correction of any electrolyte abnormalities. A further deadly complication of DKA in the pediatric population is cerebral edema. While the incidence is quite rare, the mortality rate in this condition is disproportionately higher. Because of this, once a provider identifies cerebral edema, the patient must be treated quickly with either mannitol or hypertonic saline. HHS occurs less frequently than DKA but is associated with a higher mortality rate. These patients will require aggressive fluid resuscitation, since they often are profoundly dehydrated at presentation. Additional treatment strategies include repletion of electrolyte deficits and low dose continuous insulin administration.
Endocrinology should be consulted for admission for all patients with DKA and HHS. Depending on severity, these patients often require admission to the intensive care unit. Additionally, patients newly diagnosed with DM often require admission for diabetic education. Patients with a known history of T1 DM or T2 DM who simply present with hyperglycemia can be treated in the emergency department and discharged home with a sick day plan and proper outpatient follow-up.
REFERENCES
- Hamman RF, Bell RA, Dabelea D, et al. The SEARCH for diabetes in youth study: Rationale, findings, and future directions. Diabetes Care 2014;37:3336-3344.
- Paschou SA, Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. On type 1 diabetes mellitus pathogenesis. Endocr Connect 2018;7:R38-R46.
- Mayer-Davis EJ, Kahkoska AR, Jefferies C, et al. ISPAD Clinical Practice Consensus Guidelines 2018: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes 2018;19:7-19.
- Centers for Disease Control Prevention. National Diabetes Statistics Report, 2017.
- Mayer-Davis EJ, Lawrence JM, Dabelea D, et al. Incidence trends of type 1 and type 2 diabetes among youths, 2002-2012. N Engl J Med 2017;376:1419-1429.
- Yeung WCG, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011;342:d35.
- Regnell SE, Lernmark Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017;60:1370-1381.
- Rodriguez-Calvo T. Enterovirus infection and type 1 diabetes: Unraveling the crime scene. Clin Exp Immunol 2019;195:15-24.
- Kakleas K, Soldatou A, Karachaliou F, Karavanaki K. Associated autoimmune diseases in children and adolescents with type 1 diabetes mellitus (T1DM). Autoimmun Rev 2015;14:781-797.
- Kawasaki E. Type 1 diabetes and autoimmunity. Clin Pediatr Endocrinol 2014;23: 99-105.
- Ziegler AG, Rewers M, Simell O, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013;309:2473-2479.
- American Diabetes Association. Type 2 diabetes in children and adolescents. Pediatrics 2000;105:671-680.
- Reinehr T. Type 2 diabetes mellitus in children and adolescents. World J Diabetes 2013;4:270-281.
- Chobot A, Górowska-Kowolik K, Sokołowska M, Jarosz-Chobot P. Obesity and diabetes-Not only a simple link between two epidemics. Diabetes Metab Res Rev 2018;34:e3042.
- Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: Epidemiology and treatment. Curr Diab Rep 2014;14:508.
- Umpierrez GE, Latif KA, Murphy MB, et al. Thyroid dysfunction in patients with type 1 diabetes: A longitudinal study. Diabetes Care 2003;26:1181-1185.
- Cohn A, Sofia AM, Kupfer SS. Type 1 diabetes and celiac disease: Clinical overlap and new insights into disease pathogenesis. Curr Diab Rep 2014;14:517.
- Husebye ES, Anderson MS. Autoimmune polyendocrine syndromes: Clues to type 1 diabetes pathogenesis. Immunity 2010;32:479-487.
- Shin JA, Lee JH, Lim SY, et al. Metabolic syndrome as a predictor of type 2 diabetes, and its clinical interpretations and usefulness. J Diabetes Investig 2013;4:334-343.
- Weiss R, Bremer AA, Lustig RH. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci 2013;1281: 123-140.
- Fazeli Farsani S, Souverein PC, van der Vorst MM, et al. Chronic comorbidities in children with type 1 diabetes: A population-based cohort study. Arch Dis Child 2015;100(8):763-768.
- Buchberger B, Huppertz H, Krabbe L, et al. Symptoms of depression and anxiety in youth with type 1 diabetes: A systematic review and meta-analysis. Psychoneuroendocrinology 2016;70:70-84.
- Rechenberg K, Whittemore R, Grey M. Anxiety in youth with type 1 diabetes. J Pediatr Nurs 2017;32:64-71.
- Naicker K, Johnson JA, Skogen JC, et al. Type 2 diabetes and comorbid symptoms of depression and anxiety: Longitudinal associations with mortality risk. Diabetes Care 2017;40:352-358.
- Rosenbloom AL, Silverstein J, Amemiya S, Zeitler P, Maahs DM, Klingensmith G. Type 2 Diabetes.; 2011.
- Copeland KC, Zeitler P, Geffner M, et al. Characteristics of adolescents and youth with recent-onset type 2 diabetes: The TODAY cohort at baseline. J Clin Endocrinol Metab 2011;96:159-167.
- Centers for Disease Control and Prevention. Diabetes Death Rates Among Youths Aged ≤ 19 Years — United States, 1968-2009. MMWR Morb Mortal Wkly Rep 2012;61:869-872.
- Rosenbloom AL. The management of diabetic ketoacidosis in children. Diabetes Ther 2010;1:103-120. doi:10.1007/s13300-010-0008-2
- Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis. Pediatr Diabetes 2007;8:28-43. doi:10.1111/j.1399-5448.2007.00224.x
- Fedorovich SV, Voronina PP, Waseem TV. Ketogenic diet versus ketoacidosis: What determines the influence of ketone bodies on neurons? Neural Regen Res 2018;13:2060-2063.
- Olivieri L, Chasm R. Diabetic ketoacidosis in the pediatric emergency department. Emerg Med Clin North Am 2013;31:755-773.
- Laffel L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 1999;15:412-426.
- Ozougwu JC, Obimba KC, Belonwu CD, Unakalamba CB. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J Physiol Pathophysiol 2013;4: 46-57.
- Sanyoura M, Philipson LH, Naylor R. Monogenic diabetes in children and adolescents: Recognition and treatment options. Curr Diab Rep 2018;18:58.
- ISPAD & IDF. Global IDF/ISPAD for Diabetes in Childhood and Adolescence; 2011.
- Lemelman MB, Letourneau L, Greeley SAW. Neonatal diabetes mellitus: An update on diagnosis and management. Clin Perinatol 2018;45:41-59.
- Craig ME, Jefferies C, Dabelea D, et al. ISPAD Clinical Practice Consensus Guidelines 2014. Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes 2014;15(Suppl 20):4-17.
- Repaske DR. Medication-induced diabetes mellitus. Pediatr Diabetes 2016;17:392-397.
- Zeitler P, Fu J, Tandon N, et al. ISPAD Clinical Practice Consensus Guidelines 2014. Type 2 diabetes in the child and adolescent. Pediatr Diabetes 2014;15(Suppl 20):26-46.
- Skyler JS, Bakris GL, Bonifacio E, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 2017;66:241-255.
- Soto-Rivera CL, Asaro LA, Agus MS, Decourcey DD. Suspected cerebral edema in diabetic ketoacidosis: Is there still a role for head CT in treatment decisions? Pediatr Crit Care Med 2017;18:207-212.
- Bui H, To T, Stein R, et al. Is diabetic ketoacidosis at disease onset a result of missed diagnosis? J Pediatr 2010;156:472-477.
- Wolfsdorf JI, Allgrove J, Craig ME, et al. ISPAD Clinical Practice Consensus Guidelines 2014. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. Pediatr Diabetes 2014;15(Suppl 20):154-179.
- Dunger DB, Sperling MA, Acerini CL, et al. European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics 2004;113:e133-e140.
- Yared Z, Chiasson JL. Ketoacidosis and the hyperosmolar hyperglycemic state in adult diabetic patients. Diagnosis and treatment. Minerva Med 2003;94:409-418.
- Rosenbloom AL. Hyperglycemic hyperosmolar state: An emerging pediatric problem. J Pediatr 2010;156:180-184.
- Morales AE, Rosenbloom AL. Death caused by hyperglycemic hyperosmolar state at the onset of type 2 diabetes. J Pediatr 2004;144:270-273.
- Maletkovic J, Drexler A. Diabetic ketoacidosis and hyperglycemic hyperosmolar state. Endocrinol Metab Clin North Am 2013;42:677-695.
- American Diabetes Association. 13. Children and adolescents: Standards of medical care in diabetes-2019. Diabetes Care 2019;42(Suppl 1):S148-S164.
- Long B, Koyfman A. Emergency medicine myths: Cerebral edema in pediatric diabetic ketoacidosis and intravenous fluids. J Emerg Med 2017;53:212-221.
- Glaser NS, Ghetti S, Casper TC, et al; Pediatric Emergency Care Applied Research Network (PECARN) DKA FLUID Study Group. Pediatric diabetic ketoacidosis, fluid therapy, and cerebral injury: The design of a factorial randomized controlled trial. Pediatr Diabetes 2013;14:435-446.
- Hsia DS, Tarai SG, Alimi A, et al. Fluid management in pediatric patients with DKA and rates of suspected clinical cerebral edema. Pediatr Diabetes 2015;16:338-344.
- Kuppermann N, Ghetti S, Schunk JE, et al. Clinical trial of fluid infusion rates for pediatric diabetic ketoacidosis. N Engl J Med 2018;378:2275-2287.
- Glaser N, Kuppermann N. The evaluation and management of children with diabetic ketoacidosis in the emergency department. Pediatr Emerg Care 2004;20:477-481.
- Glaser N. Cerebral injury and cerebral edema in children with diabetic ketoacidosis: Could cerebral ischemia and reperfusion injury be involved? Pediatr Diabetes 2009;10:534-541.
- Muir AB, Quisling RG, Yang MC, Rosenbloom AL. Cerebral edema in childhood diabetic ketoacidosis: Natural history, radiographic findings, and early identification. Diabetes Care 2004;27:1541-1546.
- Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema development in children with diabetic ketoacidosis. N Engl J Med 2001;344:264-269.
- Takaya J, Ohashi R, Harada Y, et al. Cerebral edema in a child with diabetic ketoacidosis before initial treatment. Pediatr Int 2007;49:395-396.
- Tiwari LK, Muralindharan J, Singhi S. Risk factors for cerebral edema in diabetic ketoacidosis in a developing country: Role of fluid refractory shock. Pediatr Crit Care Med 2012;13:e91-e96.
- Glaser NS, Wootton-Gorges SL, Buonocore MH, et al. Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr Diabetes 2006;7:75-80.
- Jones T; ISPAD Hypoglycemia Guidelines Writing Group. Defining relevant hypoglycemia measures in children and adolescents with type 1 diabetes. Pediatr Diabetes 2018;19:354-355.
- Trachtenbarg DE. Diabetic ketoacidosis. Am Fam Physician 2005;71:1705-1714.
Emergency medicine providers commonly will encounter children with type 1 and type 2 diabetes. Unfortunately, the incidence of both is increasing, and the acute care provider must be able to recognize the subtle and dramatic presentations of both diseases. Early recognition and management of both the disease and its complications — diabetic ketoacidosis, hyperglycemic hyperosmolar state, and cerebral edema — are critical to ensure an optimal outcome.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.