Central Nervous System Manifestations of Drug Toxicity
Authors: Wilfredo Rivera, MD, Toxicology Fellow, Assistant Instructor, University of Texas Southwestern Medical Center, Dallas; Rebeca Gracia, PharmD, Toxicology Fellow, North Texas Poison Center, Dallas; and Larissa I. Velez, MD, Assistant Professor of Emergency Medicine, University of Texas Southwestern Medical Center, Dallas; Associate Medical Director, North Texas Poison Center.
Peer Reviewers: Sandra M. Schneider, MD, FACEP, Professor and Chair, Department of Emergency Medicine, University of Rochester, NY; and Steven M. Winograd, MD, FACEP, Attending Physician, Emergency Department, Uniontown Hospital, Uniontown, PA.
Emergency medicine physicians routinely manage patients with neurologic toxicity due to drugs and chemicals. The causes of these toxicities are diverse. In the year 2001, there were more than 2 million human exposures reported to the American Association of Poison Control Centers Toxic Exposure Surveillance System. Of these, 498,524 patients were treated in a health care facility.1 The outcome for these patients depends on the timely identification and management of treatable causes of central nervous system (CNS) toxicity. The importance of knowing how to manage these exposures in emergency departments (EDs) cannot be overemphasized.
The manifestations of CNS toxicity have a very broad differential diagnosis, including metabolic derangements, structural lesions, psychiatric conditions, or even combinations of these. Excluding trauma and infectious etiologies is an integral part of the initial evaluation. The management of patients with evidence of CNS toxicity, thus, can be very complex and challenging. The focus of this article will be on the manifestations of drugs in the CNS, along with management recommendations.—The Editor
General Concepts
The nervous system is divided into the central and peripheral (PNS) nervous systems. The blood-brain barrier and the blood-nerve barrier have different permeability characteristics and thus are not affected equally by toxins. This accessibility variation will, in turn, determine the clinical manifestations seen in each patient.2 Some toxins may affect the PNS and some may affect just the CNS. Botulinum toxin, for example, acts at the neuromuscular junction, resulting in paralysis. In contrast, cocaine acts directly in the CNS. Some agents, like the organophosphates, which block acetylcholinesterase at all neuronal synapses, can affect both the PNS and CNS.
The CNS capacity for repair and regeneration is very limited. If the damage produced by the toxin is structural, then there will be a limited ability to return to the original level of functioning. Also, not all areas of the CNS have the same susceptibility to the effects of toxins.2 The brain is extremely sensitive to the effects of hypoxia and hypoglycemia due to its increased energy demands and limited ability to utilize alternative energy sources.
The CNS effects of drugs and chemicals can be divided into drugs that cause altered mental status, seizures, encephalopathy and movement disorders. Some indirect effects of drugs and toxins also will be discussed; these include cerebrovascular accidents and CNS infections.
Manifestations of CNS Toxicity
The clinical features of all these presentations may vary among patients and depend on the degree of intoxication and any other underlying disease processes. Toxicity from multiple agents, as with co-ingestants, can influence and complicate the clinical presentation. Some toxin combinations, for example ethanol and benzodiazepines, may have additive or synergistic effects. Other combinations, such as cocaine and opiates, will have opposing effects. The final clinical manifestation may be a complicated combination of multiple factors.
Altered Mental Status. Alterations in mental status range from agitation to coma and also can include variations in thought process, such as delirium and dementia. Many drugs can produce effects on both ends of the level of consciousness spectrum. For example, patients with anticholinergic delirium or cocaine toxicity may present with agitation or sedation. Alterations of mental status result from the imbalance between excitatory and inhibitory neurotransmitters in the brain. The main inhibitory neurotransmitter of the CNS is gamma aminobutyric acid (GABA) and the main excitatory neurotransmitter is glutamate. The catecholamines and other neurotransmitters such as serotonin also frequently are involved. Toxins can affect the neurotransmitters via changes in production, release, degradation, reuptake, and receptor activity. Agitation occurs when there is excessive neuronal excitation, as with the sympathomimetics. It also can be the result of decreased neuronal inhibition, as seen with benzodiazepine withdrawal.
Agitation. Patients can present with different levels of increased mental status, ranging from nervousness to violence. These patients may develop life-threatening complications as a result of this excessive motor activity. Rhabdomyolysis resulting from breakdown of muscle tissue is a common complication of prolonged agitation and can lead to renal failure as myoglobin precipitates and occludes kidney tubules.3-5 Agitated patients should be treated emergently, as they may pose a risk to other patients and staff.6,7
Delirium and dementia must be differentiated because they are associated with distinct differential diagnoses. Delirium is an acute organic brain syndrome with a fluctuating global cognitive impairment, attention abnormalities, altered level of consciousness, altered psychomotor activity, and/or a disordered sleep-wake cycle.8 Delirium usually is reversible and treatable. In contrast, dementia is a chronic, gradual deterioration of cognitive functions that is not associated with altered level of consciousness and does not have a fluctuating course. It often is not reversible or treatable.9
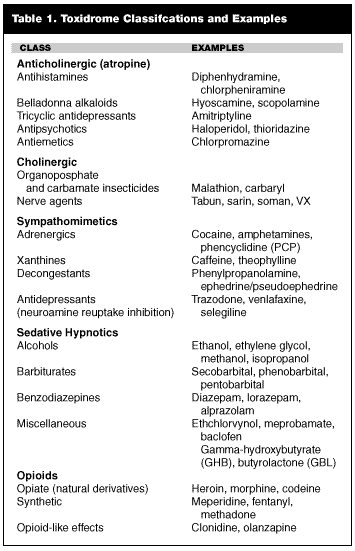
Many toxins result in delirium, and they often are grouped based upon the specific toxidrome they cause. (See Table 1.) These toxidromes include the sympathomimetic (adrenergic stimulants such as cocaine and the amphetamines), and the anticholinergic (such as atropine). Tables 1 and 2 list the toxidromes and some common causative agents. Some agents, such as ethanol and benzodiazepines, that predominately cause CNS depression, also can cause agitation as part of their withdrawal syndromes. Drug interactions that result in the serotonin syndrome and the neuroleptic malignant syndrome also can result in agitation.10-12 The psychedelics and hallucinogens, with lysergic acid diethylamide (LSD) and phencyclidine (PCP) being the classic examples, affect perception, often resulting in agitation and/or delirium. The patient’s perception of reality is distorted, with vivid illusions and heightened sensorium.13,14

Decreased Mental Status. Presenting symptoms of decreased mental status can range from drowsiness to coma. Physicians describe the levels of alertness in many different ways, sometimes leading to confusion among consultants. The following descriptions aid in the correct description of the mental status. Obtundation refers to mild to moderate reduction in alertness. Stuporous patients can be aroused only by vigorous stimulation. Comatose patients remain unarousable to any stimuli.15
The differential for decreased mental status is extensive and includes a multitude of drugs. These agents include sedative-hypnotics, opioids, barbiturates, anticonvulsants, and anticholinergics. Most drugs produce CNS depression by three pharmacological mechanisms: stimulation of GABA receptors; stimulation of opioid receptors; and stimulation of central muscarinic receptors. Catecholamine depletion also is a postulated mechanism for CNS depression, specifically in cocaine abusers.16-18 This phenomenon is referred to as the "cocaine washout syndrome." It is described as CNS depression occurring after a cocaine binge. In addition, two proposed structural mechanisms mediate decreased levels of consciousness: bilateral injury to the cerebral cortex and damage to the reticular activating system within the brain stem.15,19
Seizures. Drugs may result in seizures as part of their direct toxicity or due to a withdrawal syndrome. A common mnemonic for the differential of toxin-induced seizures is "OTIS CAMPBEL." (See Table 3.) Some of the most frequently seen causes of toxin-induced seizures include cocaine toxicity and alcohol withdrawal.20,21 Another example of withdrawal seizures is evidenced by the multiple case reports of seizures after the use of the benzodiazepine antagonist flumazenil.22-25

Toxin-induced seizures are caused by imbalances between glutamate and GABA and are mostly generalized tonic-clonic in nature.2,267 Drugs such as tricyclic antidepressants act in the sodium channel with the net effect of lowering the seizure threshold and increasing seizure potential.26 Other drugs, like cyanide, cause seizures by affecting the oxygen utilization by the brain. This results in cellular hypoxia, membrane leakage, and neuronal firing.27-29 There is evidence that adenosine is an important mediator in the termination of seizures. Adenosine seems to block the release of excitatory neurotransmitters. It also inhibits the excitatory neurotransmitter’s action at the post-synaptic membrane. These are two negative feedback mechanisms that terminate seizures.30,31 Any agent that blocks these adenosine receptors, like theophylline and caffeine, will precipitate intractable seizures.32,33
Encephalopathy. Encephalopathy is referred to as a syndrome of personality changes, decreased mental status, and diminished intellectual capacity. The direct mechanisms that produce encephalopathy are poorly understood. Drug-associated encephalopathy generally results from damage outside the CNS. For example, acetaminophen elicits liver damage, resulting in encephalopathy by the production of the toxic metabolite N-acetyl-para-quinone-imine (NAPQI) via Cytocrome P 450 (CYP2E1).34 In a recent multicenter trial of 17 tertiary care centers, acetaminophen was found to be the most common cause of acute liver failure.35 Carbon tetrachloride causes hepatocellular necrosis via the same CYP2E1 enzyme as acetaminophen.36 Amatoxins from the Amanita species of hepatotoxic mushrooms inhibit protein synthesis by binding to RNA polymerase II.37,38 The anticonvulsant valproic acid is thought to inhibit free fatty acid synthesis, resulting in microvascular steatosis, which causes accumulation of ammonia and encephalopathy.39 Thiamine deficiency can result in Wernicke’s encephalopathy due to thiamine’s critical role in the pyruvate dehydrogenase complex in the Kreb’s cycle.40-43 The inhalation of heroin vapor known as "chasing the dragon" has been associated with leukoencephalopathy and elevated brain lactate levels.44 Finally, anoxic encephalopathy can result indirectly from any drug toxicity that impairs oxygen delivery to the brain.
Movement Disorders. Movement disorders, such as drug-induced Parkinsonism, dystonia, akathisia, dyskinesias and the neuroleptic malignant syndrome, classically have been associated with the use of neuroleptic drugs.45,46 Nevertheless, other drug classes also may be responsible for some of these movement disorders.47 These drugs include tricyclic antidepressants (TCAs), monoamine oxydase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and atypical antidepressants like trazodone. Symptoms may develop within hours to weeks after initiating any of these drugs.
Drug-induced Parkinsonism is indistinguishable from idiopathic Parkinson’s disease. Dystonia is a sustained muscular contraction most commonly occurring in the face, neck, or head, which is described by patients as a cramp-like sensation. Akathisia is a subjective motor restlessness that may be interpreted wrongly by physicians as anxiety, prompting an increase in the dose of the offending agent.48 Dyskinesia is a difficulty performing voluntary movements. The pharmacologic mechanisms of these movement disorders are poorly understood, but are thought to be mediated by the effects of serotonin, acetylcholine, and dopamine in the basal ganglia.47
Botulinum toxin has been used successfully in the treatment of various forms of dystonia, suggesting an excess of acetylcholine as the pathological mechanism. Dopaminergic drugs, such as bromocriptine, appear to alleviate Parkinson’s-like symptoms, and suggest that some movement disorders may be caused by diminished dopamine activity. In contrast, the movement disorder described as chorea is treated with agents like reserpine, which are aimed at reducing dopamine and serotonin. Regardless of the underlying pathologic mechanism, the treatment for most drug-induced movement disorders just requires the discontinuation of the causative agent.48
Stroke. Stroke can result as a direct effect of drugs or an indirect complication of their use. Sympathomimetics, like cocaine and the amphetamines, may cause ischemic and hemorrhagic cerebrovascular accidents.49-54 Cocaine has been associated with several of these events, but the exact mechanism of action still is unclear. Several hypothesized mechanisms include cocaine-associated vasculitis and transient hemodynamic effects mediated by the pharmacologic action of cocaine.55-60 Chronic ephedrine use may cause acute intracerebral hemorrhage and CNS vasculitis.61,62 Several deaths have been reported due to cerebral infarction, cerebral hemorrhage, and subarachnoid hemorrhage after the use of ephedrine and its related compounds.62-65 One study shows that phenylpropanolamine (PPA) was an independent risk factor for hemorrhagic stroke in women.51
Infections. CNS infections such as meningitis and intracerebral abscesses can occur as a result of intravenous drug abuse.66 These are not the direct effect of the drug, but are a consequence of using contaminated drugs and/or paraphernalia.
Patients usually have a history of fever, general malaise, or other systemic complaints. The clinical presentation also may involve headache, seizures, alterations in the mental status, and/or signs of focality. Symptoms of the drug toxicity initially may mask the signs and symptoms of an underlying infection, making the diagnosis of a CNS infection difficult. The incidence of neurologic complications in patients with infective endocarditis varies from 20-40%, making this a very important diagnosis in the intravenous drug abuser population.66 Cerebral embolism is one of the most common CNS presentations in patients with infective endocarditis.66
History and Physical Examination
The history and physical examination are particularly important in the field of toxicology because they are used to identify toxidromes. The toxidromes often aid in narrowing the differential diagnosis and help guide management decisions. Toxidromes are general diagnostic categories based on vital signs, pupil size, bowel sounds, mental status, and skin changes. Table 2 describes the most common toxidromes along with treatment recommendations. Historical information that can influence management decisions includes a complete history of the ingestion (substance, amount, time of ingestion, and location of event), accessible medications, and past medical and psychiatric histories.
Differential Diagnosis
The differential diagnosis of CNS toxicity is very broad, including neurologic (structural), infectious, metabolic, and psychiatric (functional) etiologies, or a combination of one or more of these groups. (See Table 4.) Figure 1 shows an algorithm for the diagnosis and management of alterations in mental status.
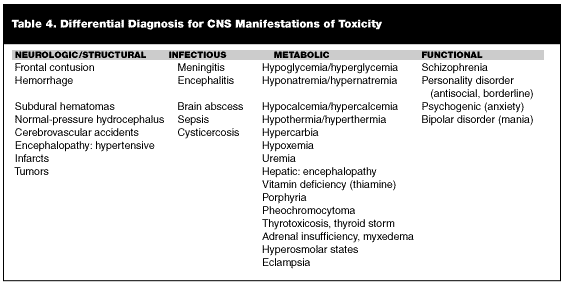
Neurologic. Any space-occupying lesion can result in alterations in mental status. Differentiation between these and toxic or metabolic alterations can be aided by assessing a few specific findings: the pupil size and response; the ocular position at rest and in response to stimulus; and the motor response to stimulus. Structural lesions will result in asymmetric or focal neurologic findings. Toxic-metabolic influences will tend to result in a dissociation of findings. In general, the pupillary reflexes seem to be relatively protected from toxic-metabolic changes, and therefore will remain intact. In cases of toxic insults to the CNS, the patients also tend to improve over time, in contrast to structural injuries.15
Metabolic/Electrolyte. Metabolic causes of altered mental status may occur independently of a toxicological cause, as well as being a complication of the toxic effects of the agent. For example, lithium toxicity can cause nephrogenic diabetes insipidus and worsening hypernatremia, eventually leading to compromised membrane stability and neuronal death. The hypernatremia manifests clinically as altered mental status. Life-threatening issues that require immediate attention include hypoxemia, commonly resulting from shock or respiratory failure, and hypoglycemia. Profound thiamine depletion has been the precipitating cause for Wernicke’s encephalopathy.67 Wernicke’s encephalopathy is referred to as the triad of ataxia, ophthalmoplegia, and altered mental status.
Functional Psychiatric. A psychiatric (functional) etiology always is a diagnosis of exclusion. Patients with catatonia, psychosis, hallucinations, or conversion disorders can present with apparent alterations of the mental status.
General Treatments
Although the specific management is different depending on the causal agent, several general interventions are important for the treatment of these patients. Meticulous attention to airway, breathing, and circulation (ABCs) and aggressive supportive management are the only interventions needed for most cases of toxicity.68-73
A useful mnemonic for common treatable causes of altered mental status is the DONT, representing D-stick or Dextrose, Oxygen, Naloxone, and Thiamine. Hypoglycemia and hypoxia must be identified and corrected immediately. The administration of naloxone will reverse opioid-induced respiratory depression.74-78 Thiamine prevents Wernicke’s encephalopathy, and should be given to all malnourished patients.67
The electrocardiogram (ECG) is a diagnostic tool that aids in the treatment decisions for some drugs. Cyclic antidepressants, massive diphenhydramine overdoses, or some anti-convulsants may cause sodium channel blockade, which will manifest as a widening of the QRS complex on the ECG.79-81 Several of the newer and older antipsychotics can cause a long QT, which can lead to torsades de pointes and death.82,83
Specific Treatments
Agitation. Agitated patients must be restrained. Physical restraints must be followed quickly by chemical (pharmacologic) restraints. This approach prevents the patient from developing multiple complications such as hyperthermia, rhabdomyolysis, and sudden death.3,84 Hobble or hog-tie restraints without any chemical restraints have been associated with sudden death in patients with excited delirium after cocaine use.3 Benzodiazepines commonly are preferred first-line agents. These drugs are easy to administer and titrate, have a fast onset of action, and have relatively few side effects.85 In extremely agitated patients, repeated doses or even the addition of other pharmacologic agents may be necessary.
Butyrophenone neuroleptics also may be used for the control of agitation. Studies comparing parenteral haloperidol vs. parenteral lorazepam86 and droperidol vs. lorazepam6 have demonstrated similar efficacy but a superior safety profile of the benzodiazepines.87 These studies focus on populations with known psychotic disorders, which may bias these studies toward a positive effect of the butyrophenones. Butyrophenone neuroleptics can predispose patients to hyperthermia, as they impair the body’s ability to exchange heat via dopaminergic blockade in the striatum or by directly altering central thermoregulatory centers.88,89 Animal studies have shown increased incidence of seizures with the use of neuroleptics.90 Cases of fatal dysrhythmias and torsades de pointes associated with droperidol have raised additional concerns.91 For this reason, we recommend the use of benzodiazepines in the patient with an unknown cause of agitation.
Patients with agitation not controlled by high doses of benzodiazepines may need the addition of barbiturates and, ultimately, nonbenzodiazepine sedating agents like propofol. The treatment of severe withdrawal syndromes, like that seen with gamma hydroxybutirate (GHB), is improved by adding barbiturates to the benzodiazepines.92 Propofol is a sedating agent with similar action to the benzodiazepines. Its main side effect is hypotension.93
Newer neuroleptics have been developed with a promising safety profile for the treatment of agitation of known psychotic origin. Ziprasidone was observed to be effective in reducing the symptoms of agitation in patients with psychosis.94,95 The study also showed that ziprasidone was well tolerated and did not result in movement disorders.94
Physostigmine is a specific antidote for the agitated delirium caused by anticholinergic agents. Physostigmine is a tertiary amine carbamate. This structure allows for CNS penetration. Administration of 1-2 mg of physostigmine over two minutes will reverse the CNS effects of the anticholinergics. The patient will become alert and awake and able to give a good history. This will eliminate costly tests and make the definitive diagnosis. Physostigmine has been associated with seizures and death in patients who overdosed with cyclic antidepressants.80 For this reason, it is essential to do an ECG before administration of physostigmine. The confirmation that the QRS width is less than 100 milliseconds makes cyclic antidepressant toxicity unlikely.
Sedation or CNS Depression. Airway management is a key concept in the treatment of these patients. One element to consider before intubation is the availability of effective antidotes to reverse opiate-induced respiratory depression. The use of these antidotes often will obviate the need for intubation. Naltrexone, nalmefene, and naloxone are competitive opioid antagonists and act at the mu, kappa, and delta receptors. Naloxone is the antagonist of choice for the treatment of opioid-induced respiratory depression. Its onset of action is one minute when given intravenously and its clinical effects usually last 45-70 minutes.96 Routes of administration for naloxone include intravenous, intramuscular, and subcutaneous. The goal of therapy with naloxone is to correct respiratory depression. The initial dose in life-threatening toxicity in adults is 2 mg intravenously. If there is no response, the dose can be repeated every three minutes, up to 10 mg. Synthetic opioids like meperidine may require doses up to 10 mg. If there is no response after 10 mg, it is highly unlikely that the manifestations are due to isolated opioid toxicity. In non-life-threatening situations, administer 0.4 mg intravenously and titrate the dose to the patient’s response. The goal of treatment is reversing respiratory depression without precipitating withdrawal in opioid-dependent patients. The duration of action of most opiates is longer than that of naloxone. For this reason, a continuous infusion may be necessary in patients requiring repeated doses of naloxone. To calculate the infusion rate, identify the response dose (dose needed for reversal of respiratory depression) and infuse two-thirds of this dose every hour as a continuous infusion. Table 5 lists common antidotes and their doses. Give one-half the response dose before starting the infusion. Tramadol is a weak mu-opioid receptor agonist and has been associated with seizures. The use of naloxone in tramadol overdoses is contraindicated.97,98

Flumazenil is an effective benzodiazepine receptor antagonist, but generally is contraindicated in the overdose setting due to the risk of precipitating withdrawal seizures. Benzodiazepine-dependent patients have decreased seizure threshold due to down regulation of GABA receptors and decreased GABA activity. The addition of a competitive benzodiazepine antagonist may potentiate withdrawal and seizures. Flumazenil also can increase seizure risk by reversing protection of benzodiazepines in the setting of other co-ingestants that can result in seizures, like cyclic antidepressants. In the setting of conscious sedation or with a pediatric patient, with a pure benzodiazepine overdose in the benzodiazepine naïve patient, no real seizure risk exists to complicate the administration of flumazenil.
The most common dose of flumazenil is 0.1-0.3 mg IV over 30 seconds. Its onset of action is 1-2 minutes. The pediatric dose is 10-20 mcg/kg IV or rectally. Flumazenil has a short half-life, and repeated dose up to 2-5 mg, or even a continuous infusion, may be required. If therapy fails even after repeated high doses, the depressive effects most likely stem from other sedative-hypnotic agents. Utilize small doses, such as 0.05 mg, of flumazenil for a benzodiazepine-dependant patient if the option of mechanical ventilation is not feasible. In these rare cases where intubation would be particularly difficult, slowly titrate flumazenil to avoid precipitating a life-threatening withdrawal.99
Seizures. Toxin-induced seizures tend to be short and self-limited. Benzodiazepines, most commonly lorazepam, are the treatment of choice. The mechanism of action of the various benzodiazepines is similar but the pharmacokinetics properties differ.100 One comparative study showed that diazepam distributed into the brain within 10 seconds and maintained EEG seizure control for 20-30 minutes, whereas lorazepam distributed into the brain within three minutes and maintained EEG seizure control for up to three hours.100,101 If after aggressive benzodiazepine administration the patient continues with seizures, barbiturates should be started as second-line agents. Phenytoin is not useful in toxin-induced seizures and even has been shown in animal models to be detrimental in the treatment of theophylline-induced seizures.100
Isoniazid toxicity ensues after depletion of pyridoxine (vitamin B6), a substrate needed for the conversion of glutamic acid to GABA by pyridoxal 5-phosphatase. The resultant decrease in GABA leads to seizures. Status epilepticus develops rapidly. Pyridoxine, in combination with a benzodiazepine, is the mainstay of treatment. The dose of pyridoxine is 5 g intravenously empirically or 70 mg/kg in children. Give 1 g of pyridoxine for each 1 g of isoniazid ingested, if ingestion amount is known.102 Pyridoxine also is the antidote for the hydrazines, such as monomethylhydrazine from mushrooms of the Gyromitra species.19
Conclusion
Many drugs and toxins affect the CNS. The main manifestations of drug toxicity in the CNS include altered mental status, encephalopathy, seizures, movement disorders, CNS infections, and stroke. Basic supportive management is the cornerstone for most of these conditions, aided by knowledge of antidotes, decontamination, and enhanced elimination techniques. Although the differential for all of these problems is extensive, a good history coupled with physical examination findings and selective use of the laboratory and radiology services will help reach the correct diagnosis.
References
1. Litovitz TL, Klein-Schwartz W, Rodgers GC Jr., et al. 2001 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2002;20: 391-452.
2. Gallagher EJ. Neurologic Principles. In: Goldfrank LR, Flomenbaum NE, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies, 7th ed. New York: McGraw-Hill; 2002:282-291.
3. Ruttenber AJ, Lawler-Heavner J, Yin M, et al. Fatal excited delirium following cocaine use: Epidemiologic findings provide new evidence for mechanisms of cocaine toxicity. J Forensic Sci 1997;42: 25-31.
4. Chan TC, Evans SD, Clark RF. Drug-induced hyperthermia. Crit Care Clin 1997;13:785-808.
5. Curry SC, Chang D, Connor D. Drug- and toxin-induced rhabdomyolysis. Ann Emerg Med 1989;18:1068-1084.
6. Richards JR, Derlet RW, Duncan DR. Chemical restraint for the agitated patient in the emergency department: Lorazepam versus droperidol. J Emerg Med 1998;16:567-573.
7. Young GP. The agitated patient in the emergency department. Emerg Med Clin North Am 1987;5:765-781.
8. Lipowski ZJ. Update on delirium. Psychiatr Clin North Am 1992; 15:335-346.
9. Rabins PV, Folstein MF. Delirium and dementia: Diagnostic criteria and fatality rates. Br J Psychiatry 1982;140:149-153.
10. Dannawi M. Possible serotonin syndrome after combination of buspirone and St John’s Wort. J Psychopharmacol 2002;16:401.
11. Radomski JW, Dursun SM, Reveley MA, et al. An exploratory approach to the serotonin syndrome: An update of clinical phenomenology and revised diagnostic criteria. Med Hypotheses 2000;55: 218-224.
12. Carbone JR. The neuroleptic malignant and serotonin syndromes. Emerg Med Clin North Am 2000;18:317-325, x.
13. Abraham HD, Aldridge AM, Gogia P. The psychopharmacology of hallucinogens. Neuropsychopharmacology 1996;14:285-298.
14. Leikin JB, Krantz AJ, Zell-Kanter M, et al. Clinical features and management of intoxication due to hallucinogenic drugs. Med Toxicol Adverse Drug Exp 1989;4:324-350.
15. Plum F, Posner JB. The Diagnosis of Stupor and Coma, vol 19, 3rd ed. Philadelphia: F.A. Davis Company; 1980.
16. Roberts JR, Greenberg MI. Cocaine washout syndrome. Ann Intern Med 2000;132:679-680.
17. Sporer KA, Lesser SH. Cocaine washed-out syndrome. Ann Emerg Med 1992;21:112.
18. Trabulsy ME. Cocaine washed out syndrome in a patient with acute myocardial infarction. Am J Emerg Med 1995;13:538-539.
19. Delaney KA, Kolecki P. Central nervous System Depression. In: Ford MD, Delaney KA, Ling LJ, et al, eds. Clinical Toxicology. 1st ed. Philadelphia: W.B. Saunders Company; 2001:137-145.
20. D’Onofrio G, Rathlev NK, Ulrich AS, et al. Lorazepam for the prevention of recurrent seizures related to alcohol. N Engl J Med 1999; 340:915-919.
21. Guerot E, Sanchez O, Diehl JL, et al. Acute complications in cocaine users. Ann Med Interne [French] 2002;153(3 Suppl): 1S27-1S31.
22. Marchant B, Wray R, Leach A, et al. Flumazenil causing convulsions and ventricular tachycardia. BMJ 1989;299:860.
23. Amrein R, Leishman B, Bentzinger C, et al. Flumazenil in benzodiazepine antagonism. Actions and clinical use in intoxications and anaesthesiology. Med Toxicol Adverse Drug Exp 1987;2:411-429.
24. Burr W, Sandham P, Judd A. Death after flumazepil. BMJ 1989; 298:1713.
25. Mordel A, Winkler E, Almog S, et al. Seizures after flumazenil administration in a case of combined benzodiazepine and tricyclic antidepressant overdose. Crit Care Med 1992;20:1733-1734.
26. Pentel PR, Keyler DE. Cyclic Antidepressants. In: Ford MD, Delaney KA, Ling LJ, et al, eds. Clinical Toxicology, 1st ed. Philadelphia: W.B. Saunders Company; 2001:515-521.
27. Bitner RS, Kanthasamy A, Isom GE, et al. Seizures and selective CA-1 hippocampal lesions induced by an excitotoxic cyanide metabolite, 2-iminothiazolidine-4-carboxylic acid. Neurotoxicology 1995;16:115-122.
28. Yamamoto H. A hypothesis for cyanide-induced tonic seizures with supporting evidence. Toxicology 1995;95:19-26.
29. KernsII W, Isom G, Kirk MA. Cyanide & Hydrogen Sulfide. In: Goldfrank LR, ed. Goldfrank’s Toxicologic Emergencies, 7th ed. New York: McGraw-Hill Medical Pub. Division; 2002:1498-1510.
30. Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 2001;24:31-55.
31. Huber A, Guttinger M, Mohler H, et al. Seizure suppression by adenosine A(2A) receptor activation in a rat model of audiogenic brainstem epilepsy. Neurosci Lett 2002;329:289-292.
32. Dragunow M, Goddard GV, Laverty R. Is adenosine an endogenous anticonvulsant? Epilepsia 1985;26:480-487.
33. Eldridge FL, Paydarfar D, Scott SC, et al. Role of endogenous adenosine in recurrent generalized seizures. Exp Neurol 1989;103: 179-185.
34. Ruepp SU, Tonge RP, Shaw J, et al. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci 2002;65:135-150.
35. Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947-954.
36. Zangar RC, Benson JM, Burnett VL, et al. Cytochrome P450 2E1 is the primary enzyme responsible for low-dose carbon tetrachloride metabolism in human liver microsomes. Chem Biol Interact 2000; 125:233-243.
37. Paaso B, Harrison DC. A new look at an old problem: Mushroom poisoning. Clinical presentations and new therapeutic approaches. Am J Med 1975;58:505-509.
38. Shi Y, He J, Chen S, et al. MARS: Optimistic therapy method in fulminant hepatic failure secondary to cytotoxic mushroom poisoning—A case report. Liver 2002;22 Suppl 2:78-80.
39. Perucca E. Pharmacological and therapeutic properties of valproate: A summary after 35 years of clinical experience. CNS Drugs 2002; 16:695-714.
40. Nakada T, Knight RT. Alcohol and the central nervous system. Med Clin North Am 1984;68:121-131.
41. Lee WM. Drug-induced hepatotoxicity. N Engl J Med 1995;333: 1118-1127.
42. Zimmerman HJ, Ishak KG. Valproate-induced hepatic injury: Analyses of 23 fatal cases. Hepatology 1982;2:591-597.
43. Otten EJ, Prybys KM, Bethgesell L. Ethanol. In: Ford MD, Delaney KA, Ling LJ, et al, eds. Clinical Toxicology, 1st ed. Philadelphia: W.B. Saunders Company; 2001:605-612.
44. Kriegstein AR, Shungu DC, Millar WS, et al. Leukoencephalopathy and raised brain lactate from heroin vapor inhalation ("chasing the dragon"). Neurology 1999;53:1765-1773.
45. Solomons K. Quetiapine and neuroleptic malignant syndrome. Can J Psychiatry 2002;47:791.
46. Stanfield SC, Privette T. Neuroleptic malignant syndrome associated with olanzapine therapy: A case report. J Emerg Med 2000;19: 355-357.
47. Gill HS, DeVane CL, Risch SC. Extrapyramidal symptoms associated with cyclic antidepressant treatment: A review of the literature and consolidating hypotheses. J Clin Psychopharmacol 1997;17: 377-389.
48. Ganzini L, Casey DE, Hoffman WF, et al. The prevalence of metoclopramide-induced tardive dyskinesia and acute extrapyramidal movement disorders. Arch Intern Med 1993;153:1469-1475.
49. Rowbotham MC. Neurologic aspects of cocaine abuse. West J Med 1988;149:442-448.
50. Kalant H. The pharmacology and toxicology of "ecstasy" (MDMA) and related drugs. CMAJ 2001;165:917-928.
51. Kernan WN, Viscoli CM, Brass LM, et al. Phenylpropanolamine and the risk of hemorrhagic stroke. N Engl J Med 2000;343: 1826-1832.
52. Perez JA Jr., Arsura EL, Strategos S. Methamphetamine-related stroke: Four cases. J Emerg Med 1999;17:469-471.
53. Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4-methylenedioxymethamphetamine ("ecstasy"). Lancet 1992;340: 384-387.
54. Shukla D. Intracranial hemorrhage associated with amphetamine use. Neurology 1982;32:917-918.
55. Nolte KB, Brass LM, Fletterick CF. Intracranial hemorrhage associated with cocaine abuse: A prospective autopsy study. Neurology 1996;46:1291-1296.
56. Satel SL, Gawin FH. Migrainelike headache and cocaine use. JAMA 1989;261:2995-2996.
57. Levine SR, Brust JC, Futrell N, et al. Cerebrovascular complications of the use of the "crack" form of alkaloidal cocaine. N Engl J Med 1990;323:699-704.
58. Mody CK, Miller BL, McIntyre HB, et al. Neurologic complications of cocaine abuse. Neurology 1988;38:1189-1193.
59. Singhal AB, Caviness VS, Begleiter AF, et al. Cerebral vasoconstriction and stroke after use of serotonergic drugs. Neurology 2002;58:130-133.
60. Qureshi AI, Suri MF, Guterman LR, et al. Cocaine use and the likelihood of nonfatal myocardial infarction and stroke: Data from the Third National Health and Nutrition Examination Survey. Circulation 2001;103:502-506.
61. Yin PA. Ephedrine-induced intracerebral hemorrhage and central nervous system vasculitis. Stroke 1990;21:1641.
62. Buxton N, McConachie NS. Amphetamine abuse and intracranial haemorrhage. J R Soc Med 2000;93:472-477.
63. Agaba EA, Lynch RM, Baskaran A, et al. Massive intracerebral hematoma and extradural hematoma in amphetamine abuse. Am J Emerg Med 2002;20:55-57.
64. Moriya F, Hashimoto Y. A case of fatal hemorrhage in the cerebral ventricles following intravenous use of methamphetamine. Forensic Sci Int 2002;129:104-109.
65. McEvoy AW, Kitchen ND, Thomas DG. Intracerebral haemorrhage and drug abuse in young adults. Br J Neurosurg 2000;14:449-454.
66. Tunkel AR, Pradhan SK. Central nervous system infections in injection drug users. Infect Dis Clin North Am 2002;16:589-605.
67. Hoffman RS, Goldfrank LR. The poisoned patient with altered consciousness. Controversies in the use of a "coma cocktail." JAMA 1995;274:562-569.
68. Vale JA. Position statement: Gastric lavage. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35:711-719.
69. Chyka PA, Seger D. Position statement: Single-dose activated charcoal. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35:721-741.
701. Krenzelok EP, McGuigan M, Lheur P. Position statement: Ipecac syrup. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35:699-709.
71. Barceloux D, McGuigan M, Hartigan-Go K. Position statement: Cathartics. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35:743-752.
72. Tenenbein M. Position statement: Whole bowel irrigation. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35: 753-762.
73. Bond GR. The role of activated charcoal and gastric emptying in gastrointestinal decontamination: A state-of-the-art review. Ann Emerg Med 2002;39:273-286.
74. Gaddis GM, Watson WA. Naloxone-associated patient violence: An overlooked toxicity? Ann Pharmacother 1992;26:196-198.
75. Moore RA, Rumack BH, Conner CS, et al. Naloxone: Underdosage after narcotic poisoning. Am J Dis Child 1980;134:156-158.
76. Evans LE, Swainson CP, Roscoe P, et al. Treatment of drug overdosage with naloxone, a specific narcotic antagonist. Lancet 1973;1: 452-455.
77. Kulig K, Duffy J, Rumack BH, et al. Naloxone for treatment of clonidine overdose. JAMA 1982;247:1697.
78. North DS, Wieland MJ, Peterson CD, et al. Naloxone administration in clonidine overdosage. Ann Emerg Med 1981;10:397.
79. Yang YC, Kuo CC. Inhibition of Na(+) current by imipramine and related compounds: Different binding kinetics as an inactivation stabilizer and as an open channel blocker. Mol Pharmacol 2002;62: 1228-1237.
80. Boehnert MT, Lovejoy FH, Jr. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. N Engl J Med 1985;313:474-479.
81. Brown TC, Barker GA, Dunlop ME, et al. The use of sodium bicarbonate in the treatment of tricyclic antidepressant-induced arrhythmias. Anaesth Intensive Care 1973;1:203-210.
82. Brice JH, Pirrallo RG, Racht E, et al. Management of the violent patient. Prehosp Emerg Care 2003;7:48-55.
83. Rao KA, Adlakha A, Verma-Ansil B, et al. Torsades de pointes ventricular tachycardia associated with overdose of astemizole. Mayo Clin Proc 1994;69:589-593.
84. Stratton SJ, Rogers C, Green K. Sudden death in individuals in hobble restraints during paramedic transport. Ann Emerg Med 1995;25: 710-712.
85. Dietch JT, Jennings RK. Aggressive dyscontrol in patients treated with benzodiazepines. J Clin Psychiatry 1988;49:184-188.
86. Salzman C, Solomon D, Miyawaki E, et al. Parenteral lorazepam versus parenteral haloperidol for the control of psychotic disruptive behavior. J Clin Psychiatry 1991;52:177-180.
87. Thomas H, Jr., Schwartz E, Petrilli R. Droperidol versus haloperidol for chemical restraint of agitated and combative patients. Ann Emerg Med 1992;21:407-413.
88. Heiman-Patterson TD. Neuroleptic malignant syndrome and malignant hyperthermia. Important issues for the medical consultant. Med Clin North Am 1993;77:477-492.
89. Susi U. Vassallo KAD. Thermoregulatory Principles. In: Goldfrank LR, ed. Goldfrank’s Toxicologic Emergencies, 7th ed. New York London: McGraw-Hill Medical Pub. Division; 2002:261-281.
90. Yokota K, Tatebayashi H, Matsuo T, et al. The effects of neuroleptics on the GABA-induced Cl- current in rat dorsal root ganglion neurons: Differences between some neuroleptics. Br J Pharmacol 2002;135:1547-1555.
91. Bailey P, Norton R, Karan S. The FDA droperidol warning: Is it justified? Anesthesiology 2002;97:288-289.
92. Dyer JE, Roth B, Hyma BA. Gamma-hydroxybutyrate withdrawal syndrome. Ann Emerg Med 2001;37:147-153.
93. Velez-Daubon L, Delaney KA. Central Nervous System Agitation. In: Ford MD, Delaney KA, Ling LJ, Erickson T, eds. Clinical Toxicology, 1st ed. Philadelphia: W.B. Saunders Company; 2001: 146-153.
94. Daniel DG, Potkin SG, Reeves KR, et al. Intramuscular (IM) ziprasidone 20 mg is effective in reducing acute agitation associated with psychosis: A double-blind, randomized trial. Psychopharmacology (Berl) 2001;155:128-134.
95. Citrome L, Volavka J. Violent patients in the emergency setting. Psychiatr Clin North Am 1999;22:789-801.
96. Chamberlain JM, Klein BL. A comprehensive review of naloxone for the emergency physician. Am J Emerg Med 1994;12:650-660.
97. Tobias JD. Seizure after overdose of tramadol. South Med J 1997; 90:826-827.
98. Gardner JS, Blough D, Drinkard CR, et al. Tramadol and seizures: A surveillance study in a managed care population. Pharmacotherapy 2000;20:1423-1431.
99. Weinbroum AA, Flaishon R, Sorkine P, et al. A risk-benefit assessment of flumazenil in the management of benzodiazepine overdose. Drug Saf 1997;17:181-196.
100. Bey TA, Walters FG. Seizures. In: Ford MD, Delaney KA, Ling LJ, et al, eds. Clinical Toxicology, 1st ed. Philadelphia: W.B. Saunders Company; 2001:155-167.
101. Dundee JW, McGowan WA, Lilburn JK, et al. Comparison of the actions of diazepam and lorazepam. Br J Anaesth 1979;51:439-446.
102. Sievers ML, Herrier RN. Treatment of acute isoniazid toxicity. Am J Hosp Pharm 1975;32:202-206.
Emergency medicine physicians routinely manage patients with neurologic toxicity due to drugs and chemicals. The causes of these toxicities are diverse. The focus of this article will be on the manifestations of drugs in the CNS, along with management recommendations.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.