Chemical Warfare Agents, Part I: Choking Agents, Vesicants, and Halogenated Oximes
Part I: Choking Agents, Vesicants, and Halogenated Oximes
Author: Charles E. Stewart, MD, FACEP, Emergency Physician, Colorado Springs, CO.
Peer Reviewers: Cynthia K. Aaron, MD, FACMT, FACEP, Associate Professor of Emergency Medicine, Director, Clinical Toxicology Services, University of Massachusetts Memorial Medical Center, Worcester, MA; Jonathan L. Burstein, MD, FACEP, Director of Disaster Medicine, Beth Israel Deaconess Medical Center; Assistant Professor of Medicine, Harvard Medical School, Boston, MA.
The spread of anthrax through the postal system in 2001 brought home the threat of bioterrorism. Now, the current war with Iraq and concerns about potential terrorism make chemical warfare agents and weapons of mass destruction important concerns. The terrorists involved in attacks of Sept. 11, 2001, and others have studied in-depth the possibilities of using chemical agents to wreak havoc on the innocent. For these terrorists, the possibility of chemical weapons is close at hand. In 1997, Secretary of Defense William Cohen identified Libya, Iraq, Iran, and Syria as countries that aggressively were seeking nuclear, biological, and chemical weapons. Indeed, Iraq and Iran already have used chemical weapons against each other and against the Kurdish population.
Chemical warfare is not a popular topic, but the potential of chemical warfare agents should be of overwhelming concern to civilian emergency physicians and prehospital providers. As General Pershing warned after World War I, "the effect is so deadly to the unprepared that we can never afford to neglect the question."
What are the predominant agents? What injuries do they cause? What is the prognosis for their victims? These are questions that both military and civilian physicians alike should understand. Part I of this series will cover choking agents, vesicants, and halogenated oximes. Part II will address nerve agents and blood agents.—The Editor
Terrorist Actions
It is quite unlikely that the civilian emergency physician will see these chemical warfare agents employed in wartime. More important to the civilian physician and the poison control center is the possibility that these agents might be employed by a terrorist group. Even though the military has control of the agents in most countries, it is quite possible that terrorist groups could gain access to these agents. It also is possible that a "state" sponsor could supply some terrorist groups with these weapons. Chemical warfare agents easily are synthesized from readily available chemicals, and terrorist operations, such as Aum Shinriko, can simply make their own.
The recent use of toxic chemical agents in the Tokyo and other Japanese subways mandate preparedness by civilian medical providers. In addition, chemical plants recently have been listed as potential targets for terrorist attacks. The chemical tragedy that killed more than 2,000 people at Bhopal, India, in 1984 should convince any reluctant emergency physician that preparedness is necessary, even when no terrorists are involved.
The Civilian Threats
Although a wide range of chemical weapons has been used in prior conflicts, only a small number of these compounds developed by the military is likely to be used by an aggressor or terrorist. With a high volatility, compounds such as phosgene, chlorine, hydrogen sulfide, and hydrogen cyanide could be used in a confined area such as a subway or building. Mustard gas and nerve agents have a high enough lethality and could be employed in a small enough package to interest the terrorist. In proper form, these agents can persist for long periods and could deny use of airports, water supplies, bridges, or even highways.
These agents often are either production chemicals or first derivatives of chemicals used to produce plastics, pesticides, and fabrics. Modern pesticide-producing plants can manufacture nerve agents, while ethylene and sulfurated petrochemicals may be combined in the Levinstein process to produce sulfur mustard gas.1
Equally likely is the use of precursors or industrial chemicals as improvised chemical warfare agents. There is a wide range of chemicals that the military considers "unsuitable" for use as a chemical warfare agent that have significant application as agents of terrorism. Releases of ammonia and chlorine in the civilian world have forced evacuation of hundreds of families in recent times.2 The devastation of Bhopal, India, from the accidental release of toxic methyl isocyanate gas on an unprepared civilian population has been well chronicled. With the exception of chlorine’s early use in World War I, none of these gases has a military application or designation.
An act of sabotage on a tanker or tank truck in the downtown area of a middle-American city rapidly could discharge tons of potentially lethal vapors. Few firefighters and hazardous materials crews are trained to mitigate the sudden, deliberate release of tons of vapor by explosive devices, accompanied by potential booby-trapping and subsequent secondary explosions. If prevailing winds were favorable to the terrorists, the tragedy at Bhopal could be brought to the United States.
The local evacuation, mitigation, and decontamination of both the scene and the casualties are well beyond the scope of an article to introduce their medical treatment by emergency physicians. Unfortunately, if the sarin releases in Japan are any guide, the casualties from a chemical release often will present directly to the emergency department (ED). Decontamination of the casualty may be a requirement, unless it already has been done. This must be addressed in another article, but some agent-specific decontamination guidance is provided here.
Some Terms
Chemical agents may exist in solid, liquid, or gaseous form, depending on the temperature. Most military chemical agents are dispersed as gases, liquids, or an aerosol (very small, solid particles or liquid droplets suspended in a gas). Tear gas, for example, is not a gas at all, but an aerosolized solid. Mustard "gas" also is not a gas, but a liquid that may or may not evaporate (again, depending on the temperature). Remember that a vapor is the gaseous form of a substance that exists at a lower temperature than the boiling point of the gas.
Another aspect of chemical agent classification is whether a particular chemical is persistent or non-persistent. Persistency is used to describe how long an area remains contaminated at a level of toxicity dangerous to humans after a chemical agent has been employed. Thus, an agent is called persistent if the area in which it was used remains contaminated for about a day or more. Some persistent chemical agents, such as mustard and VX, do not evaporate easily and tend to prevent the use of terrain or property until they are dispersed or decontaminated.
Volatility and persistence are inversely related. A substance is more volatile if it evaporates quickly. A non-persistent or volatile agent disperses quickly, sometimes in a matter of minutes or hours. Lewisite, cyanide, ammonia, chlorine, tearing agents, and sarin are non-persistent agents. Non-persistent agents present less contamination danger to emergency medical service (EMS) or medical providers in the hospital. (These generalizations are subject to temperature, wind, and some surface characteristics.)
To make a non-persistent agent persistent, thickeners can be added. As a result of the additional chemical additive, it becomes more complicated to destroy the agent by certain means. In general, these agents will present significant dangers to the EMS providers at the scene and may well cause contamination of ambulances and even EDs. The more persistent an agent is, the more decontamination is required.
A few specific terms familiar to the military chemist should be noted. LD50 is well known to emergency physicians and represents the dose of a drug that will be fatal to 50% of the population. A counterpart exists for incapacitating agents: ID50. The incapacitating effects will be felt by 50% of the population exposed to an ID50. This concept could be extended to cover any effect of an agent: ED50 represents the effective dose for 50% of the population.
Since the concept of dose applies only to injected, absorbed, or ingested chemicals, another term was devised for inhalation agents. This is the concentration time product or C•t, often simply expressed as Ct. C•t refers to the agent concentration (usually in mg/m3) multiplied by the time of exposure in minutes. Exposure of a concentration of 10 mg/m3 of tabun for five minutes results in a C•t of 50 mg-min/m3. Exposure of a concentration of 5 mg/m3 of tabun for 10 minutes also results in a C•t of 50 mg-min/m3. For almost all chemical vapors and gases, the C•t associated with a specific biological effect is relatively constant even though the concentration and time may vary (within some limits). This means that a 10-minute exposure to 5 mg-min/m3, a 5-minute exposure to 10 mg-min/m3, and a one-minute exposure to 50 mg-min/m3 will all have about the same effect on the unprotected patient.
The inhaled agent’s ECt50, ICt50, and LCt50 correspond to the ingested/injected drug’s ED50, ID50, and LD50, respectively. The C•t does not take respiratory rate and depth into account. The exercising soldier will have a different exposure than the sedentary patient. This difference may be significant at lower concentrations, but at high concentrations, there may be little difference between the two subjects because of the rapidity with which the effective CT50 is achieved in both.
Lethal Agents
Although any chemical spray or gas may have lethal potential, any listing of lethal chemical agents usually includes choking (pulmonary) agents, vesicants, nerve agents, and cyanide. (See Table 1.) These agents may be used to destroy and demoralize the civilian population or to deny an area of operations to the populace or military.

Choking (Pulmonary) Agents
Chemical agents that attack lung tissue, primarily causing pulmonary edema, are classed as lung-damaging agents. The choking agent or pulmonary agents are characterized by pronounced irritation of the upper and lower respiratory tract.
This group contains phosgene (CG), diphosgene (DP), chlorine (Cl), and chloropicrin (PS). Inhalation of organohalides, oxides of nitrogen, and many other compounds can cause the same symptoms as chlorine and phosgene. Perfluoroisobutylene (PFIB) is a toxic pyrolysis product of Teflon and other tetrafluoroethylene polymers encountered in civilian and military construction. The oxides of nitrogen are used in industry, components of blast weapons, or may be toxic decomposition products. Smoke-producing devices may contain toxic compounds that cause the same effects as phosgene. The medical management of phosgene and chlorine exposure also applies to casualties from compounds such as PFIB or nitrogen oxides.
These agents are treacherous because they often have a latent period following exposure. Chemically induced acute lung injury by these agents involves a permeability defect in the blood-air interface (the alveolar-capillary membrane). A victim with dyspnea and mild chest discomfort may progress in severity over a few hours after exposure.
Chlorine. Chlorine is a slightly water-soluble, yellowish gas that is about 2.5 times heavier than air. The use of 498 tons of chlorine released from 20,730 cylinders on April 22, 1915, was the cause of more than 7000 immediate casualties and about 10,000 more casualties treated during the ensuing few days at Ypres, Belgium.3 The troops were totally unprotected from this agent. This agent has fallen into military disfavor because it is quite easy to detect, dissipates quickly, and is relatively easy to protect against.
It is readily available as an improvisational weapon for terrorists. Millions of tons of chlorine are used in bleaching, water purification, chemical processes, and swimming pools. It is probably the cause of more cases of accidental industrial toxic exposure than any other single agent.4
Adding chlorine bleach to an acidic cleaning agent will produce free chlorine gas. The extent of the resulting injury depends on the concentration and duration of the exposure. Symptoms begin in moments, and no delayed symptoms are noted. Clinical effects are noted in the table.
Chlorine gas is heavier than air and may be delivered by bomb, artillery, or mortar round, or simply released from a canister. It follows the curves of the terrain and will flow into basements, subways, caves, trenches, and hollows. During World War I in Ypres, Belgium, the German military released the agent from pressurized canisters, creating a 5-foot-high cloud that extended four miles.3
Chlorine forms both hydrochloric (HCL) and hypochlorous acid on contact with water. Hypochlorous acid spontaneously will decompose to hypochloric acid and free oxygen radicals. Free chlorine and all of the combination products of chlorine and water will disrupt cellular proteins. Molecular chlorine is about 10-30 times more toxic than the combination products.5
The severity of injury caused by chlorine gas is related to the concentration of the gas, the duration of exposure (the C•t product as described earlier), and the water content of the tissues exposed. (See Table 2.) Chlorine will react with water to form hypochlorite and hydrochloric acid, which are extremely irritating to respiratory tissues. Exposure to chlorine gas at several parts per million will cause eye and throat irritation within minutes. High-level exposure of several hundred to several thousand parts per million will cause intense burning of the eyes, nose, and throat, followed shortly thereafter by coughing, shortness of breath, production of whitish sputum, and substernal pain. Severe exposure may result in pulmonary edema, hypoxemia, and respiratory failure.

Chlorine effects usually are related to the extent of pulmonary damage. The typical pulmonary pattern of those exposed to a moderate concentration of chlorine gas is an increased airway resistance with normal diffusion capacity. Pulmonary function tests will return to normal in most patients within months.6 As might be expected, patients with worse subjective complaints tended to have slower resolution than those with simpler complaints, such as cough.7
Most victims exposed to chlorine gas will recover without problems. Only if the patient has exposure to high concentrations of chlorine would significant sequelae be found. Prolonged pulmonary disease after exposure to chlorine is not common.
Decontamination. Decontamination of the victim should begin with rapid removal of clothing and then copious irrigation of irritated areas. After removal of the patient’s clothing and irrigation, the medical provider needs no special contamination protection for pure chlorine exposure.
Specific Treatment. Oxygen. Measure the oxygen saturation in all symptomatic patients, since hypoxia is common. These patients frequently need oxygen supplementation. Humidified oxygen should be continued until symptoms abate. Early administration of intermittent positive pressure breathing, positive end expiratory pressure, or bilevel positive airway pressure (BiPAP) may be quite helpful in minimizing the pulmonary edema and reducing the degree of hypoxia.8 Intubation with or without a ventilator may be appropriate and should be guided by clinical judgment.
Nebulizer Treatments. Patients with hyperactive airways may benefit from aerosolized bronchodilators such as albuterol.9 Beta-adrenergic agonists relax airway smooth muscle and reduce hyperactivity and resultant airway narrowing in patients with chlorine inhalation. Aminophylline, theophylline, or terbutaline may be considered in these patients.10
Multiple authors have recommended nebulized sodium bicarbonate for treatment of inhaled chlorine gas. Symptomatic treatment for chlorine inhalation may include nebulized inhalation of a mixture of 2 mL 8.4% NaHCO3 with 2 mL normal saline.11-13 It is thought to neutralize the hydrochloric acid produced in the alveoli. These recommendations are based on anecdotal evidence and may or may not be effective. There is controversy, and some authors feel bicarbonate inhalation still is inadequately supported by evidence.9,14 Nebulized lidocaine may provide analgesia and reduce coughing.
Steroids. Administration of steroids has been recommended, but there is no good proof of their beneficial effects in the treatment of pulmonary edema due to phosgene inhalation.15 When steroids are used, they should be given in high doses by inhalation or intravenously.
Antibiotics. Antimicrobial therapy should be reserved for a proven bacterial bronchitis/pneumonia. Bacterial superinfection resulting in bronchitis or pneumonia may present 3-5 days after exposure to chlorine. The clinician should search for this infection if the patient does not improve within 3-4 days after exposure to chlorine. Prophylactic therapy is not appropriate.16
Rest. It is important that a patient exposed to chlorine be kept at rest until the danger of pulmonary edema is past. This may not be possible in some operational situations, but if the patient has significant respiratory involvement, litter evacuation is necessary.
Eye injuries should be evaluated with fluorescein staining under magnification, preferably with a slit lamp.
Diuretics. The pulmonary edema associated with chlorine is noncardiac. Treatment of the chlorine-induced noncardiac pulmonary edema is supportive. Diuretics such as furosemide have only a very limited utility in these patients and generally should not be used.
Intubation and positive end expiratory pressure may reduce the pulmonary edema.
Threat. Use of chlorine gas in an unconfined area would require massive amounts of chlorine to be released in a short period of time. This would be a daunting undertaking for a terrorist. Chlorine could be effective within a closed environment such as a subway or auditorium. Since other agents are readily available, chlorine is an unlikely terrorist weapon, but the emergency provider needs to be well aware of chlorine’s toxicity and treatment, since accidental exposures to chlorine are quite common.
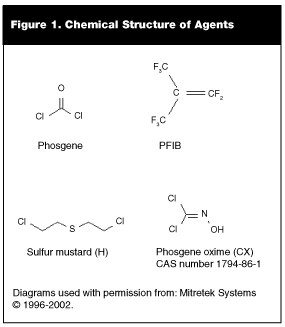
Phosgene. Phosgene (COCl2 or carbonyl chloride) is a chemical intermediate in the production of plastics, dyes, and insecticides. (For chemical structure, see Figure 1.) Phosgene artillery shells used by Germany in 1917 in Verdun were the first battlefield use of the agent.
Phosgene is a colorless gas under ordinary conditions of temperature and pressure. Its boiling point is 8.2°C, making it an extremely volatile and non-persistent agent. The military designator for this agent is CG, and the similar and related chemical diphosgene is designated as DP. Phosgene easily is liquefied under pressure and is transported as a liquid.
Organochlorine compounds may decompose to phosgene when heated. This includes polyvinyl chloride plastics, Teflon, and carbon tetrachloride. Phosgene should be considered a product of combustion and may be found in most house fires.
Phosgene is a chemical agent used to cause pulmonary damage and asphyxia. Although phosgene is a liquid, it rapidly vaporizes to a colorless, low-lying gas that is three times as dense as air. It has a characteristic odor of new-mown hay, but this is not a sufficient warning to prevent toxicity. It does not cause immediate damage and, thus, often has a lengthy and deep exposure. It is one of the most deadly of the chemical war munitions because of this latent period. The British used hand grenades with chlorine and phosgene in World War I, while the Germans used phosgene shells.17
The average lethal dose is 500-800 ppm/min. This means that a 10-minute exposure to only 50-80 ppm likely would be lethal. The military defines a concentration of phosgene of 2 ppm as immediately dangerous to life and health (IDLH).18 Mild symptoms (i.e., eye, nose, and throat irritation) occur at 3-5 ppm. Pulmonary edema occurs at doses exceeding 600 mg/min/m3. During World War I, phosgene was responsible for more than 60-80% of the "gas" deaths. The fact that phosgene is less potent than almost all of the subsequently developed chemical warfare agents should not lead to an underestimation of its danger; deaths have occurred after only a few breaths of this gas.
Mechanism of Action. The mechanism of action of phosgene is not fully understood. Theories for the action include the dissolution of phosgene within the alveolus to hydrochloric acid and subsequent alveolar damage, inhibition of enzymes, and a direct reaction at the alveolar and capillary wall interface.
Phosgene is only slightly soluble in water and aqueous solutions. Once dissolved, it rapidly hydrolyzes to form carbon dioxide and hydrochloric acid. The early effects of phosgene are due to its decomposition in the presence of water within the mucous membranes to form carbon dioxide and hydrochloric acid. The early onset of ocular, nasal, pharyngeal, and central airway irritation is caused by this decomposition.
Late effects appear to occur from acylation reactions with amino, hydroxyl, and sulfhydryl groups in the alveoli and the capillaries in the lung.19
Whatever the mechanism of action, phosgene increases the permeability of the alveolar capillaries, allowing plasma to flood the alveoli and create pulmonary edema. This leads to leakage of fluid from the capillaries within the alveolus. This leakage increases over 24 hours, leading to clinically evident pulmonary edema. The effects reach a maximum at about 12-24 hours after exposure. As higher doses are inhaled, the latent period becomes shorter.
Clinical Effects. Initial exposure to low concentration can cause burning and watering of the eyes, pharyngitis, dry cough, chest tightness, and dyspnea. (See Table 3.) Nausea and headache also may be present. These symptoms occur during and immediately after the exposure, but clear within a short time. The presence or absence of these symptoms may be of little value in the ultimate prognosis.20

At dosages exceeding 120 mg-min/m3 (30 ppm/min) the initial respiratory symptoms are followed by a second (latent) phase lasting 24-48 hours.21 Duration of this latent phase is inversely proportional to the inhaled dose. A small dose may have a latent phase of 24-48 hours or may have no further symptoms. A large inhaled dose may be followed by additional symptoms within 1-4 hours after exposure.
The outstanding feature of phosgene intoxication is a third phase: massive pulmonary edema that is identical to the pulmonary edema found in adult respiratory distress syndrome. After a latent period of about 2-24 hours as described above, pulmonary edema develops, with production of large amounts of frothy sputum.22 The patient may have an initial cough, followed by dyspnea; rapid, shallow breathing; and cyanosis. As the edema progresses, discomfort, apprehension, dyspnea, and cyanosis increase, and the patient develops the characteristic frothy sputum. The patient may have rales and rhonchi or diminished breath sounds. This pulmonary edema does not respond to furosemide, intravenous nitrates, or morphine. As noted above, the exact site of action has not been determined.19
The most prominent symptom that occurs after the latent period is dyspnea. This may be accompanied by chest tightness. Dyspnea reflects the increasing accumulation of interstitial pulmonary fluid, decreased lung compliance, and increased ventilatory drive. The clinician may hear fine crackles and rales, first in the bases, then in the entire lung field.
The trachea and bronchi usually are normal in appearance. This is in contrast with chlorine and chloropicrin poisoning, as discussed later in this article. At high concentrations, there may be some erythema of the oral and pharyngeal mucosa.
Severe mucous membrane irritation associated with phosgene occurs from a high-level exposure and indicates serious toxicity. With exposures to very high concentrations of phosgene, death may occur within hours. In most cases, the pulmonary edema maximizes in 24 hours. If the patient survives for 48 hours, there is a very good prognosis and, in the absence of complicating infections, the patient may have little or no residual damage. Residual bronchitis may last for several days.
Extensive fluid loss into the lungs may cause hypovolemic shock. Phosgene also may cause vasoconstriction within the lungs.
Although the major determinant of the pulmonary damage is the inhaled dose of phosgene, increased physical activity will shorten the latent period and increase the respiratory distress. Very large inhalation doses of phosgene can cause laryngeal irritation, laryngeal spasm, and death from this mechanism.
At very high concentration, phosgene will pass through the alveolar-capillary membrane and cause hemolysis in the pulmonary capillaries. The red cell fragments may block the capillary circulation. Death occurs in these victims within a few minutes from acute cor pulmonale.
Symptomatic patients should have a chest x-ray to assess the possibility of noncardiogenic pulmonary edema. Common radiologic abnormalities include diffuse interstitial infiltrates, normal cardiac shadow, and possibly a bilateral perihilar fluffy infiltrate. These radiologic changes frequently lag behind the clinical presentation in phosgene inhalation. If the patient becomes worse, then a repeat radiograph is appropriate.
Arterial blood gases will show a metabolic acidosis with a compensatory hyperventilation.23 The gases also will show hypoxia.
Hypoxia may be accompanied by hypotension as vast quantities of fluid are coughed up from the lungs. Death results from respiratory failure, hypoxia, hypovolemia, or a combination of these. Rapid progression of hypoxia and hypotension suggest a poor prognosis. If the patient develops pulmonary edema within four hours of exposure, the patient has a poor prognosis. Unless there is intensive medical care immediately available, these patients are at very high risk of death.
The terminal clinical phase of lethal phosgene poisoning is described as extreme distress with intolerable dyspnea until the respiration ceases.
Most survivors of acute exposure to phosgene will have a good prognosis. Shortness of breath and reduced tolerance for physical activity may persist in some patients. Smoking appears to worsen the chances of full recovery. Pre-existing lung disease, such as emphysema, will exacerbate the effects of phosgene exposure.24 Late complications include infections and slow respiratory failure.
Decontamination. Phosgene will not remain in the liquid form unless the temperature is nearly freezing. Indeed, exposure to liquid phosgene can cause cold injury or frostbite. Decontamination stations should be well-ventilated, and much of the decontamination is accomplished by simple aeration. Further decontamination will not be required unless phosgene has been used in a very cold climate.
Phosgene decomposes to carbon monoxide and hydrochloric acid in the presence of moisture. Decontamination with copious amounts of water sufficiently will decompose any residual of this agent.
Phosgene and similar pulmonary agents are absorbed by inhalation. They do not have dermal activity. Removal of clothing will prevent outgassing.
The activated charcoal in the canister of the military chemical protective mask adsorbs phosgene, and the military mask provides full protection from this gas. Various civilian protective masks may or may not provide protection. Medical providers in decontamination stations should be protected with charcoal filter respirators as a minimum.
Specific Treatment. There is no specific antidote for inhalation of phosgene. Since there is a distinct latent period associated with inhalation of phosgene, it is important to observe these patients for a minimum of six hours after exposure. Onset of pulmonary edema before six hours is predictive of severe inhalation injury.
Oxygen and Similar Therapy. Measure the oxygen saturation in all symptomatic patients, since hypoxia is common. These patients frequently need oxygen supplementation. Early administration of intermittent positive pressure breathing, positive end expiratory pressure, or BiPAP may be quite helpful in minimizing the pulmonary edema and reducing the degree of hypoxia. Intubation with or without a ventilator may be appropriate and should be guided by clinical judgment.20,25,26
Patients with hyperactive airways may benefit from aerosolized bronchodilators such as albuterol.9 Beta-adrenergic agonists relax airway smooth muscle and reduce hyperactivity and resultant airway narrowing in patients with phosgene inhalation. Aminophylline may be considered in these patients.10
Steroids. Administration of steroids has been recommended, but there is no solid proof of their beneficial effects in the treatment of pulmonary edema due to phosgene inhalation. Prednisolone has been used to prevent pulmonary edema in the asymptomatic phase after inhalation of the phosgene. A dose of 250 mg intravenously is recommended. A dose of 1 gram intravenously has been recommended for the treatment of phosgene-induced pulmonary edema.27 There is insufficient experience to judge whether these recommendations have any validity, but there are few side effects to single-dose steroid administration.
Antibiotics. Antimicrobial therapy should be reserved for a proven bacterial bronchitis/pneumonia. Prophylactic therapy is not appropriate.
Rest. It is important that a patient exposed to phosgene be kept at rest until the danger of pulmonary edema is past. This may not be possible in some operational situations, but if the patient has significant respiratory involvement, litter evacuation is necessary.
Diuretics. The pulmonary edema associated with phosgene is noncardiac. Diuretics such as furosemide have only a very limited utility in these patients and generally should not be used. Since the pulmonary edema is of such scope as to cause hypovolemia, EMS providers should be aware that use of diuretics may not only be ineffective, but may cause hypotension.
Other Therapies. In animals, one or two large doses of ibuprofen decreased the toxicity of inhaled phosgene.28 An acetylcysteine aerosol given by nebulizer also has been effective in treatment of phosgene inhalation in animals. There are few potential side effects to these treatments, but they have not been studied in humans.
Detection and Laboratory Aids. Detection of phosgene by the new-mown hay odor is neither reliable nor safe. The odor threshold for phosgene is about 1.5 mg/m3, and phosgene irritates mucous membranes at about 4 mg/m3. When the mucous membranes are irritated, the sense of smell is destroyed. There is no available laboratory test for the identification of phosgene inhalation.
Chest radiographs may show hyperinflation early, followed by pulmonary edema without cardiomegaly. Early pulmonary edema may be detected by a chest x-ray using 50-80 KV exposure. Use of 100-120 KV exposure may miss these early signs.20
Peak expiratory flow (PEF) may decrease early after a massive phosgene exposure. PEF may assess the degree of airway damage and indicate how much of the bronchoconstriction is reversible with bronchodilators. Pulmonary function testing will show increased work of breathing, increased airway resistance, decreased lung compliance, and decreased flow rates in the presence of a pulmonary injury. The presence of normal spirometry values probably excludes a significant injury to the lower respiratory tract.29 Unfortunately, pulmonary function tests often are not obtainable if the patient is a young child, unconscious, or not cooperative.
Arterial blood gases show nonspecific hypoxia. The hematocrit may increase as fluid is lost within the lung.
Threat. Phosgene is a commercial precursor to plastics manufacture. The United States produces more than one billion pounds of phosgene per year for industrial uses. It is shipped in large containers in public commerce. It had a proven track record as a significant war agent in World War I. As such, it is an attractive improvised agent for terrorists.
PFIB. Perfluoroisobutylene (PFIB) is an extremely toxic pyrolysis product of Teflon and other tetrafluoroethylene polymers encountered in civilian and military construction.30 PFIB is 10 times more lethal than phosgene. (For chemical structure, see Figure 1.)
While PFIB once was used as an industrial chemical, it fell into disuse decades ago because it was so hazardous. It was prepared by the former Soviet Union as a chemical warfare agent.31
Like phosgene, it also produces pulmonary edema.
Clinical Effects. Inhalation exposure may cause symptoms of pulmonary edema with wheezing, difficulty breathing, production of sputum, and cyanosis. The patient may experience initial coughing and chest pain followed by a latent period of several hours and then rapidly decompensate.
High concentrations may produce irritation of the eyes, nose, and throat, but the major target organ of this agent is the lungs. Systemic effects seen in animal studies only occur after there is substantial injury to the lung, and hypoxia is considered a major contributing factor.
There is little data on the dosages that cause symptoms in human beings. In rodents, dosages of 150-180 ppm/min (1250-1500 mg-min/m3) will kill 50% of the test population. A comparable dose of phosgene is 750 ppm/min.32,33
High concentrations of PFIB have caused sudden death in animals. There is no such reported death in humans, although reported experience is scant.
A comparable syndrome known as "polymer fume fever" has been described following the inhalation of the products from the combustion of Teflon.34-37 These inhalation injuries have occurred from fires, smoking cigarettes contaminated with Teflon or similar products, and welding around Teflon.
A few hours after exposure, there is a gradual increase in temperature (hence the moniker "polymer fume fever"), pulse, and respiratory rate. Shivering and sweating usually follow. Auscultation of the lungs may reveal moist, diffuse rales in the most severe cases. Radiograph of the chest may show diffuse infiltration of the lung fields without cardiomegaly. Toxic pulmonary edema may be more severe and appear earlier if the patient exercises after exposure.
Decontamination and Protection. Air purifying respirators are inadequate protection for this material. PFIB is not well adsorbed to activated charcoal. The clinical worker should have a positive pressure supplied air respirator.
When dissolved in water, PFIB decomposes rapidly to various reactive intermediates and fluorophosgene, which in turn decomposes to carbon dioxide, a radical anion, and hydrogen fluoride. Since a reactive product is hydrogen fluoride, use of water decontamination for this agent cannot be recommended. Fortunately, this agent is extremely volatile, and ventilation with air should provide adequate decontamination.
Specific Treatment. There is no prophylaxis or antidote for inhalation of PFIB. Protection against the lethal effects of inhaled PFIB has been demonstrated in rates when N-acetylcysteine was given orally 4-8 hours prior to exposure to PFIB.38 There is no known post-exposure medical or chemical therapy that reverses or impedes the injury caused by PFIB.32
Since there is a distinct latent period associated with inhalation of phosgene, it is important to observe these patients for a minimum of 6 hours after exposure. Onset of pulmonary edema before six hours is predictive of severe inhalation injury.
Oxygen. Measure the oxygen saturation in all symptomatic patients, since hypoxia is common. These patients frequently need oxygen supplementation. Early administration of intermittent positive pressure breathing, positive end expiratory pressure (PEEP), continuous positive airway pressure (CPAP), or BiPAP may be quite helpful in minimizing the pulmonary edema and reducing the degree of hypoxia. Intubation with or without a ventilator may be appropriate and should be guided by clinical judgment. Patients with hyperactive airways may benefit from aerosolized bronchodilators such as albuterol.
Steroids. Administration of steroids has been recommended but there is no good proof of their beneficial effects in the treatment of pulmonary edema due to PFIB inhalation. Indeed, there are only two reports of steroid use in PFIB inhalation in the literature—both in the same patient.36
Antibiotics. Antimicrobial therapy should be reserved for a proven bacterial bronchitis/pneumonia. Prophylactic therapy is not appropriate. Bacterial superinfection is sufficiently common to warrant a careful watch.
Rest. It is important that a patient exposed to PFIB be kept at rest until the danger of pulmonary edema is past. This may not be possible in some operational situations, but if the patient has significant respiratory involvement, litter evacuation is necessary. Pulmonary edema may be more severe and appear earlier if the patient exercises after exposure.
Diuretics. The pulmonary edema associated with PFIB is noncardiac. Diuretics such as furosemide have only a very limited utility in these patients and generally should not be used. Since the pulmonary edema is of such scope as to cause hypovolemia, EMS providers should be aware that use of diuretics may not only be ineffective, but may cause hypotension.
Fluid replacement is mandatory if the patient becomes hypotensive. Combined hypotension and hypoxia may cause multi-organ failure.
Threat. This agent is an attractive potential terrorist weapon. A charcoal-adsorbing protective mask would be inadequate for any substantial concentration of this agent. PFIB is not readily available, but Teflon is. This agent could be made easily by heating Teflon to more than 400 degrees and collecting the gas.
Fortunately, making this compound in miniscule quantities is not the same as collecting sufficient quantity at sufficient concentration and deploying it in an improvised munition. The task of collecting sufficient quantity to deploy it as an improvised munition would be orders of magnitude more difficult than making it in small quantities.
Since the former Soviet Union investigated this agent because of its ability to break through a charcoal mask, there may be quantities of it for sale on the black market.
Vesicant (Blister) Agents
A vesicant is a chemical that produces blisters or vesicles. These chemicals are blistering agents that are toxic to skin, lungs, eyes, and mucous membranes. With exposure to many of these agents, the effects noted by the victim are delayed. Blister agents are likely to be used to produce casualties and force soldiers, police, and emergency response workers to wear full protective equipment that degrades performance and efficiency.
There are three different subclasses within the vesicants: The mustards, the arsenicals (Lewisite and similar agents), and the halogenated oximes (i.e., phosgene oxime [CX]). The effects and properties of the halogenated oximes are quite different from those of the other vesicants.
There are many other compounds, such as hydrofluoric acid and fuming nitric acid, that cause similar tissue destruction, but have not been classified by the military as vesicants. Some of these chemicals will be discussed in the section on improvised agents.
Although the vesicants have a relatively low lethality, they are quite effective in inflicting painful burns and blisters that require medical attention, even at low doses. Even though these contact poisons cause serious skin and ocular lesions, pulmonary involvement is the most common cause of death.39,40 These chemicals are far less potent than nerve agents under comparable conditions of exposure.
Warfare use of vesicant gases decreases the opponent’s ability to fight by producing chemical burns on any exposed tissues or by requiring protective gear that degrades the soldier’s ability to fight. The vesicant agents would be ideal for terrorism since they are easy to make or available in industrial quantities, and produce terrible injuries in the unprotected populace. They easily could be added as a second or tertiary effect agents to endanger emergency response workers.
Mustard Agents. The mustard agents are a family of sulfur-, nitrogen-, and oxygen-based compounds with similar chemical and biological effects. The military prototype of this class of chemicals is sulfur mustard, also called Levenstein mustard after the inventor of a major manufacturing process for sulfur mustard. There are two types of sulfur mustard: distilled sulfur mustard (HD) and impure mustard (H). Other similar agents include agent Q, agent T, and the nitrogen mustards. Only sulfur mustard will be discussed in this article. (For chemical structure, see Figure 1.)
The most dangerous chemical warfare agent of World War I was sulfur mustard, and this agent still is considered a major chemical warfare threat. Iraq made extensive use of mustard against Iran in the Iran-Iraq war in the 1980s.41
History. Sulfur mustard often is called the "king" of chemical agents since it is easy to produce, inexpensive, has predictable properties, is persistent, and causes resource-devouring casualties rather than fatalities.42
The English and French named the chemical mustard after the smell or the yellowish-brown color, and Yperite after the location of the first battle where it was used. The Germans called it LOST or S-LOST after the two chemists who suggested its use as a weapon (LOmmell and STeinkopf), or yellow cross from the identifying mark on the World War I shells. Allied soldiers also called it H or HS—for "Hun Stuff," hence the NATO designation H.
Mustard most recently was employed by Iraq during the Iran-Iraq conflict of 1980-1988.41,43 One source estimates that 45,000 casualties were due to mustard.44
Although mustard was introduced late in World War I, in July of 1917, it caused more chemical casualties than all other agents combined. While lethality of the agent is low, casualties are common and numerous. In the Iran-Iraq War, many fatalities occurred because Iranian soldiers, feeling no effects, continued to wear mustard-soaked clothing and inhale its fumes.
Sulfur mustard still is considered by the military as a chemical warfare agent because of its powerful penetrating potency, persistence, and low warning properties. This combination makes personal protection very difficult. Mustard on the skin causes no immediate sensation and symptoms do not normally appear until several hours after exposure. At incapacitating levels of mustard, symptoms may not be evident for as long as 12 hours. Not only the air is dangerous; everything the mustard gas touches is potentially toxic. It is representative of the vesicant or blister agents.
Physical Characteristics. Sulfur mustard is a clear, yellow, or amber-colored oily liquid with a faint sweet odor of mustard or garlic. Mustard vaporizes slowly at temperate climates and may be aerosolized with spraying or by explosive blasts. The vapor is heavier than air and settles slowly into low areas.
Because of its low volatility at lower temperatures, mustard is considered a persistent agent at temperate climates and may remain present for up to a week after dispersal. In the desert, at temperatures above 37.7° C, mustard would be expected to persist for only a day. It is only slightly soluble in water, but highly soluble in organic solvents and in skin oils. It is hydrolyzed slowly in water to form hydrochloric acid.
Mustard freezes at 13.9° C (57° F), so it is difficult to use in the winter or to deliver by air. Mustard’s freezing point can be depressed by multiple agents, including carbon tetrachloride and Lewisite. Mustard’s high freezing point makes it quite dangerous during the times of year when the temperature falls during the night to below 10° C . The mustard will freeze in low-lying parts of the ground and then will be warmed by the sun during the day and evaporate, giving a "second attack" several hours after daybreak.
Mustard agents may be delivered by bomb, artillery, or mortar round, or by release from canisters. Missiles readily are adapted to deliver this agent in large quantities. Mustard easily is vaporized by heat and quickly spreads by wind. Mustard vapor injury is markedly enhanced by high humidity in a hot environment. It is possible to disseminate mustards that have been adsorbed by small particles ("dusty" mustard).
Mustard will go through ordinary clothing without burning it and attacks only living tissue; the reaction is manifested only after several hours have passed.45
Mechanism of Action on Tissues. Since mustard has been a popular military agent since World War I, there is a large body of literature available about the biochemical interactions of mustards. (See Table 4.) Public health considerations about the destruction and demilitarization of stocks of chemical agents have further inspired writers to research available data about mustard.

About 20% of sulfur mustard that contacts the skin is absorbed. Because mustard is lipophilic, it penetrates intact skin rapidly. Penetration is more rapid down the hair follicles and sweat glands than through intact skin. It has been estimated that between 12% and 50% of mustard that is absorbed will react with the skin and skin components. Of this, about 70% is absorbed within the epidermis and 30% within the dermis.
Mustard is a primary tissue irritant. There is no significant allergic component to the injury caused by mustard.
The molecular mechanisms that cause toxicity from mustard are far from clear.46 Sulfur mustard alkylation reactions are rapid and irreversible. These reactions cause simultaneous, multiple, complex sites of damage. It appears that mustard actively is transported across the cell membrane using a system involved in choline transport. DNA damage, inhibition of glycolysis, depletion of glutathione (used for detoxifying oxidative reactions within the cell), membrane damage, and production of cytokines all have been proposed as part of the damage produced by mustard.47 Evidence suggests that all of the proposed theories may contribute or interact to produce the cellular damage.
Mustard’s most important metabolic effect, at least for acute toxicity, is inhibition of cellular glycolysis. Disruption of that metabolic pathway is found after exposure to nearly all vesicant chemicals, usually due to interference with sulfhydryl-rich hexokinase enzyme systems.42 Mustard-mediated inhibition of glycolysis occurs as an indirect result of DNA damage and repair.
Mustard gas binds with a double alkylation reaction to DNA, forming cross-links that prevent further replication of cells. The exact site of damage is not known in all cases. Sulfur mustard does cause rapid alkylation of the purine bases (guanine and adenine) of DNA. Activation of endonucleases then leads to removal (depurination) of alkylated bases, leaving apurinic sites where DNA breaks occur readily. In turn, need for DNA repair activates poly (ADP-ribose) polymerase, an enzyme that rapidly depletes cellular NAD+.
Depletion of the cellular NAD+ takes about an hour and leads to inhibition of glycolysis, release of tissue proteases, and cellular death. The entire process takes about four hours and parallels the clinical course of the toxin.42
Mustard also damages RNA and proteins. Although mustard can react with a large variety of molecules, the dosage of mustard encountered by most patients will not inhibit the cell’s energy metabolism, protein synthesis, RNA synthesis, or other enzymatically mediated activities in any significant degree. In mammalian cells there are multiple pathways for DNA repair that may minimize the permanent damage from mustard.
Mustard is mutagenic, carcinogenic, and teratogenic. Because mustard produces lesions that are similar to the effects of x-rays, mustard gas often is termed a radiomimetic agent. Rapidly dividing cells are the most sensitive to the effects of mustard, accounting for the similarities between mustard and radiation. Although dividing cells are the most sensitive, mustard at significant concentration will kill any cells. Cells with high cell turnover such as bone marrow, intestinal mucosal cells, and skin elements are especially sensitive to mustard.
Effects of mustard exposure are cumulative if exposures occur within 12 hours.48 At more prolonged intervals between exposures, the effects probably are less accumulative due to DNA repair mechanisms and the ability of the body to replenish scavengers of reactive oxygen molecules and electrophiles.48
Symptoms. Mustard readily penetrates skin and mucous membranes. Moist body parts such as the eyes, mouth, respiratory tract, scrotum, and anus are most affected by all of the mustard agents. (See Table 5.) Between 80% and 90% of the mustard penetrates the skin and enters the circulation. Sulfur mustard affects those tissues with high replication rates, such as bone marrow and gastrointestinal tissues.

The clinical effects of mustard occur after skin, eye, or inhalation exposure. There is a characteristic latent period of 4-12 hours between exposure and the onset of symptoms. The rate of onset is related to the dose of the agent absorbed. The toxic effect of mustard depends on both the concentration of mustard in the air inhaled or skin exposed [C] and duration of exposure (t) [C•t product]. Higher concentrations and longer duration exposures cause symptoms that develop more rapidly. There also is a significant variation in the individual sensitivity to mustard.
Battlefield air concentrations during World War I mustard gas attacks were estimated in the range of 19-33 mg/m3.42 At these concentrations, exposure for several minutes causes skin and eye damage, and exposure for 30-60 minutes will cause severe respiratory injury, systemic poisoning, and probable lethality.
A 10-mcg drop applied to the skin will cause vesicle formation. Of this, about 80% will evaporate and 10% will enter the circulation. The entire vesicle, thus, is formed by only 1 mcg of mustard. A single teaspoonful of mustard over 25% of body surface area is sufficient to cause death.49
As noted previously, early recognition of vesicant agents is difficult because the initial effects may be only irritation of mucous membranes, similar to that found with either the choking agents or the tear gases. The first complaint of Iranian troops exposed to mustard gas usually was ocular: photophobia, sensation of foreign body, and conjunctivitis.50 Other presenting symptoms of these casualties were dyspnea, nausea, vomiting, headache, and fatigue. This may lead to substantial increase in the exposure, since the patient may be completely unaware of his/her danger for hours.
Victims of mustard toxicity generally have lesions in multiple sites. In most patients, lesions are primarily cutaneous, but respiratory, ocular, and gastrointestinal manifestations may occur. In One study of eight unprotected Iranian mustard victims, all had conjunctivitis, and two had serious corneal injuries. Three had respiratory distress.51
Dermal. If the patient was wearing a protective mask at the time of exposure, only cutaneous manifestations are likely. Mustard does not produce uniform skin injuries. The face, scrotum, and anal areas frequently are involved, while the thicker skin on the hands may be spared. Moist or wet skin, such as groin and axilla, is especially prone to damage; scrotal and perianal burns occurred in nearly half of U.S. survivors of World War I mustard gas attacks.52 There is no available data about perineal and vulvar involvement in females.
There are large individual differences in reactivity to sulfur mustard. As much as 100 times the agent needed to cause a lesion in a sensitive person may be required to cause similar lesions in a resistant subject.53
After a latent period of 4-12 hours, victims may develop diffuse erythema mimicking scarlet fever that progresses to blister formation and widespread bullae, similar to those of toxic epidermal necrolysis.43 The affected areas may turn black.
If the skin directly is exposed to mustard liquid, ulcerations and skin necrosis develop after only a very short latent period. Skin toxicity increases as ambient temperature increases.54
Mild cases may only have erythema similar to a sunburn. Large mustard injuries are likely to have systemic effects. The most severe cases may have marked fluid losses, hypovolemia, and renal failure.
The Iranian data presented in one study showed that 92% of 525 Iranian casualties had skin lesions, 79% had erythema, 55% had bullae, and 20% had increased pigmentation of the skin.55 Of particular note, 5% of the casualties had urticaria, and 1.2% had purpura.
These skin changes are the result of a chemical burn that heals in about 4-6 weeks unless there is an infection. After the inflammation subsides, there often is a distinctive residual black-to-brown punctate perifollicular pigmentation.56
Skin lesions in children appear to be more severe than in adults. The time of onset of symptoms is shorter when compared with adults. Both phenomena could be attributed to the more delicate skin of young patients.57
Skin lesions caused by mustard can recur at sites of prior injury. Exposed German and Japanese mustard workers had multiple skin tumors such as basal cell carcinoma, Bowen’s carcinoma, and squamous cell skin cancer.49
Ocular. In World War I, American physicians noted that the eye was the most commonly damaged organ in a mustard attack.43 The eye is affected at lower vapor concentrations than any other organ. Recovery of mild conjunctivitis takes only 1-2 weeks after exposure. Severe conjunctivitis, blepharospasm, lid edema, and conjunctival edema may be noted and may take 2-5 weeks to resolve.
Moderate corneal involvement may be noted as corneal erosion that stains with fluorescein. This may be followed by superficial corneal scarring and vascularization and iritis. These cases may take as long as 2-3 months to resolve.
If severe corneal involvement is noted, the patient may have dense corneal opacification and deep ulcer formation. These patients will require ophthalmologic intervention and may require hospitalization. Some of these patients will develop blindness due to the ocular damage.
Respiratory. Inhalation of mustard aerosol or vapor produces a serious upper respiratory tract irritation and can lead to respiratory failure and death. In World War I, the mortality of mustard gas casualties was about 2.5%, and nearly all of the deaths were from inhalation of the agent.
After a delay of several hours, victims suffer tracheobronchitis with symptoms that include chest pressure, hacking cough, sore throat, and hoarseness. Sinusitis, sinus pain, increasing cough, and tachypnea develop as symptoms progress during the next 12 hours. The severe cough responds poorly to cough suppressants, steam, bronchodilators, or corticosteroids.
The tracheal and bronchial mucosa may slough and obstruct the bronchi. Pulmonary edema is much less common with mustard gas than with the choking agents. Severe exposures lead to hemorrhagic pulmonary edema, secondary pneumonia, and respiratory failure after 24-48 hours. Bronchopneumonia is a common complication in inhalation injuries due to mustard and Lewisite. X-rays may be consistent with adult respiratory distress syndrome.
In World War I, thousands had chronic respiratory disease from exposure to mustard and phosgene. Chronic pulmonary effects expected after acute, severe mustard inhalation include chronic bronchitis reactive airway dysfunction syndrome and emphysema.58 The chronic inflammation of airways and pulmonary parenchyma can cause central stenosis, bronchiectasis, and bronchiolitis obliterans. Evidence suggests that World War I battlefield exposure resulted in a slightly increased incidence of lung cancer after a latent period of 20 years or more.59,60
Gastrointestinal. Gastrointestinal injury can result from systemic toxicity due to massive exposures (more than 1000 mg-min/m3) or swallowing contaminated food, water, or saliva. Nausea and vomiting are common after exposure; diarrhea and gastrointestinal bleeding are uncommon.61 Both the liquid and vapor forms can contaminate food, causing gastrointestinal disease.
Hematologic. High-dose exposures (more than 1000 mg-min/m3) that produce systemic toxicity may cause bone marrow suppression. The patient may develop an initial leukocytosis that lasts several days. This is followed by leukopenia. The nadir is reached at about 10 days after exposure.61 Thrombocytopenia may accompany the leukopenia. Anemia is seen less often, although all elements of the marrow are suppressed.
Cardiovascular. Large exposures to mustard can cause cardiovascular collapse, shock, and death. This may be unresponsive to fluid replacement. Patients with large skin burns have hypovo-lemia, hemoconcentration, peripheral edema, and bradycardia followed by tachycardia.
Lethality. Lethality rates after battlefield exposure are about 1-3% of mustard gas victims. The cause of death may be burns, respiratory tract damage, bone marrow depression, or infection.62 Development of mustard-induced hematological effects is an especially poor prognostic sign.
As noted previously, marked increases in the lethality will occur if the victim continues to wear mustard-soaked clothing and inhale its fumes.
Immune System. Seven to 10 days after exposure, sulfur mustard also can cause impairment of immune functions. This can increase vulnerability to bacterial infection and multiple septic complications.
Decontamination. Decontamination within 1-2 minutes after exposure is the only known effective means of preventing or decreasing tissue damage from mustard exposure. If decontamination is performed when symptoms appear, it is of only limited usefulness to the patient. It may protect medical staff from contamination and prevent spread to other parts of the body.
Protection of the medical staff is the prime priority. (In Heully and Gruninger’s description of a mustard shell explosion, 11 victims were family members and medical staff who attended the three patients.)45 Decontamination of all patients potentially exposed to mustard should be done before they are allowed into a medical treatment facility. Contaminated litters, blankets, and equipment should be left outdoors so that vapor does not accumulate indoors. The hazard from vapor is markedly higher in hot conditions than in cold weather.
Symptoms should not guide decontamination efforts. Mustard on the skin causes no immediate symptoms and symptoms may not be present for several hours after exposure.
Droplets of the oily agent should be removed by blotting and then cleansing with soap and water or a decontamination solution. Washing with 0.5% hypochlorite (household bleach diluted 1:10) is the decontamination technique most often recommended.63 In a large study of Iranian patients from the Iran-Iraq war, prolonged washing with water also was effective.55 This is controversial, and most references note that because mustard is relatively water insoluble, water alone has only limited value as a decontamination solution.64
All clothing, jewelry, and leather should be removed, and the underlying skin must be washed. Washing should include the groin, axillae, and perianal areas, since these frequently are involved. Scrubbing and hot water should be avoided, as both enhance the absorption of the agent.
Chloramine solution wash has been recommended by some military authors but generally is not available for civilians. For skin decontamination from mustard, the U.S. military currently uses M258A1 kits, which contain three sets of two paper towels each, one towel containing phenol and hydroxide, and the other containing chloramine.
If water is in short supply, a limited skin decontamination can be performed with adsorbent powders such as flour or talcum powder.65 The powder is dusted liberally onto the contaminated exposed skin, allowed to adsorb mustard from the skin, and then wiped off with a moist paper or towel. This procedure markedly will decrease contamination and subsequent exposure to the patient and staff.
Therapy. After more than 75 years of exposure to this agent, there is not yet an effective antidote for mustard. Treatment is supportive, and most patients exposed to mustard gas will recover completely.62
The primary effect of an attack with sulfur mustard is production of many patients with painful skin and eye irritation and pulmonary injuries. These wounded patients would place an enormous burden on medical services.
Mustard injuries are slow to heal and often take more than six weeks of convalescence. Only a small proportion of patients will have long-term eye or lung damage. Unfortunately, there are no studies about the incidence of cancer or birth defects following exposure to mustard.
Casualties from mustard can be classified into three categories: minimal, life-threatening, and those who require ongoing hospital care. There will be many patients with minimal injuries, the majority will require hospitalization, and a very small percentage will have life-threatening injuries.
Severe: Patients who become critically ill from exposure to mustard present with large areas of burns, major pulmonary damage, and subsequent immunosuppression. Patients with early onset of moderate-to-severe respiratory symptoms or large surface area burns caused by liquid mustard probably have life-threatening injuries. An area of erythema that covers more than 50% of the patient’s body suggests that the individual has had more than 2 LD50 of mustard exposure. Dyspnea that occurs within 4-6 hours after inhalation of mustard suggests that the patient has inhaled a lethal amount of the chemical agent. Many of these casualties will die from their exposure. As noted earlier, in the Iran-Iraq War, many fatalities occurred because Iranian soldiers continued to wear mustard-soaked clothing and inhale its fumes. The cause of death may be burns, respiratory tract damage, bone marrow depression, or infection.62
Moderate: Patients with a large area of blisters or erythema will need hospitalization. Those who have eye injuries that obscure vision or very painful eye injuries will require hospital care. Those patients with respiratory symptoms, including dyspnea or a productive cough likely will require inpatient respiratory care. Some of these patients later may progress to life-threatening symptoms. This is the largest group of patients, and those who will benefit most from medical care.
Mild: Patients with only a small area of blister or erythema will not need hospitalization. Patients with late-onset, mild upper respiratory symptoms can be treated as outpatients. Patients with conjunctivitis or mild eye irritation can be seen by an ophthalmologist in a clinic.
General. These patients will be in pain and will require significant pain medication for relief. The clinician must pay strict attention to fluid and electrolyte replacement. The patient likely will need intravenous therapy for hydration. Patients with extensive genital lesions may require a urinary catheter.
Systemic Symptoms. Systemic symptoms are uncommon in dealing with military exposure to sulfur mustard. Physicians familiar with the effects of other alkylating agents used in clinical medicine deal mainly with their systemic toxicity to cells of the bone marrow, lymphoid tissue, and gut mucosa. Exposure to sulfur mustard in wartime and terrorism involves external epithelial surfaces rather than primary systemic absorption. This probably accounts for the paucity of systemic effects seen in World War I.
Systemic symptoms have been treated with high doses of sodium thiosulfate and vitamin E. Intravenous n-acetylcysteine has been used in some patients with systemic symptoms and may have some internal decontamination effect.66 Intravenous cysteine 0.5%, 10 mg QID was used in 10 Iraqi patients in one study.62 In that same study, charcoal hemoperfusion was used in the most difficult cases.
Respiratory Therapy. Death from exposure to mustard usually is from pulmonary complications, such as acute chemical pneumonitis or pneumonia, typically exacerbated by the hematopoietic suppression.67 Appropriate respiratory care is a major contributor to survival.
The therapeutic goal in treating a patient exposed to mustard with mild respiratory symptoms is comfort management. Patients with mild upper airway lesions may benefit from cough suppressive drugs and increased humidity of the inhaled air.
Bronchodilator therapy and supplemental oxygenation are needed in all but the mildest exposures. Steroids have been used, but there is no confirmation of efficacy. Steroids should be considered in patients with underlying asthma, chronic obstructive pulmonary disease (COPD), or hyperreactive airway disease.
In patients with severe respiratory exposure, the patient must have respiratory support. If the large lower tract airways are involved, the patient may develop fever, leukocytosis, and an infiltrate on the chest x-ray during the first two days. This almost always is a chemical pneumonitis and not pneumonia, so antibiotics should be withheld. The patient subsequently may develop purulent sputum. This should be examined before the patient is started on antibiotics.
Pulmonary function tests show obstruction to airflow, and hyperinflation, as well as restrictive phenomena and progressive impairment of gas exchange.40
A patient with severe respiratory signs should have early intubation. Subsequent laryngeal spasm may make later intubation problematic. Bronchoscopy may be needed to remove secretions and plugs. Early use of positive end-expiratory pressure or constant end-expiratory pressure is beneficial. A requirement for ventilation support is ominous. More than 80% of Iranian casualties treated in Western European hospitals who required assisted ventilation died.61
Prophylactic antibiotic therapy is not recommended.
Ocular Therapy. Rapid decontamination is essential, as eye injury occurs in moments. Indeed, the military feels that the danger from exposure to the eyes is greater than the risk of exposure to vapor. The soldier is advised to remove his mask and flush the eyes out within the first few seconds after exposure.
Eye exposures may be treated with copious irrigation with 2.5% sodium thiosulfate. Minor eye lesions may be treated with soothing eye solutions. More severe eye injuries should be treated with topical antibiotics and atropine or other topical cycloplegic drugs. The patient should have frequent irrigation to remove inflammatory and necrotic debris. Topical ophthalmologic corticosteroids may be used for the first few days, but there is little data to support continued use. Topical anesthetics should be used only for examinations to prevent self-injury of the intensely inflamed eyes. Lid edge inflammation should be covered with petrolatum to ensure that lids do not adhere to each other. Dark glasses and reassurance are very important, as the eye lesions will produce severe photophobia and subsequent fear of blindness.62
To prevent complications, the patient should be treated by an ophthalmologist as soon as possible.
Skin Lesions. Chemical burns due to mustard are deceptively superficial on initial presentation. A potential lethal exposure is approximately 100 mg/kg or 5-7 mL of distilled mustard, the amount of mustard that will lightly cover about 25% of the body surface area. Patients who have sustained a potentially lethal dose of sulfur mustard may present initially with what appears to be a large first- or second-degree bum.
The blister fluid is described as non-toxic, but skin may well have residual agent present. Bullae and surrounding skin should be decontaminated carefully to remove residual agent. Large areas of denuded skin require the same care as needed for care of other serious burns.55
A major goal of skin therapy is to prevent infection. This may be accomplished by frequent irrigation of the burned areas and liberal use of topical measures, including cleansing with povidone iodine solution and application of 1% silver sulfadiazine cream.
Protection. Preprotection. Thiosulfate reacts with the cyclized form of mustard in vitro. Unfortunately, to achieve protection from the systemic effects of mustard, the antidote must be injected 10-45 minutes prior to exposure to mustard.68 Thiosulfate has no preventative effect when injected after exposure to mustard.
Free oxygen radical scavengers, copper-zinc, and manganese superoxide dismutase (SOD) were used to treat mustard skin burns in an experimental guinea pig model.69 Each of the SOD compounds dramatically reduced burn lesion area when administered intraperitoneally/intralesionally (i.p./i.l.) one hour before wound infliction. SOD had no protective effects when administered after the burn.
Other preprotection chemicals such as glutathione are being studied.70
Similar Agents. Agent T (bis[2(2-chloroethylthio)ethyl]ether) is a byproduct of one manufacturing process for making mustard gas. Very little information is available on the long-term toxicity of agent T, which has a much lower volatility than mustard. Agent T may be mixed with mustard for increased inhalation injury (this may be referred to as agent HT). For practical purposes, agents T and HT will be considered as close variants of mustard with a more rapid onset. Agent T increases the stability of the mixture so that it is more persistent.
Nitrogen mustard [methyl bis(B-chloroethyl)amine] is an oily, pale yellow to colorless liquid that is freely soluble in organic solvents but insoluble in water. Topical nitrogen mustard has been used in therapy in the treatment of mycosis fungoides.71 It never has been used in chemical warfare.
Halogenated Oximes
Phosgene oxime (CX). Phosgene oxime is an example of the class of chemical agents called urticariants or nettle gases. These agents produce an immediate sense of pain that may vary from a mild prickling to intense pain.
Introduction. Phosgene oxime, also known as CX or dichloroform oxime, is an irritant which produces erythema, wheals, and urticaria. The lesions have been compared to those caused by the stinging nettle.
CX is a white crystalline powder that melts between 39-40° C, and boils at 129° C (but it decomposes rapidly at this temperature). It is fairly soluble in water and in organic solvents. In aqueous solution, CX hydrolyses fairly rapidly, especially in the presence of alkali. It has a high vapor pressure and its odor is very unpleasant and irritating. Even as a dry solid, phosgene oxime decomposes spontaneously and has to be stored at low temperatures. By adding other chemicals, it is possible to liquefy CX at room temperature, and the liquid form is much more readily dispersed.
There is relatively little information on this agent, since it probably has not been used on a battlefield. There were indications of Iraqi use of an agent whose effects resembled CX against Iran, but there is no confirmation of this use.49,72
CX is unique because it penetrates garments and rubber much more quickly than other chemical agents do. It also acts very rapidly. It can be mixed with other chemical agents (such as VX) so that the skin damage caused by phosgene oxime will allow a second agent to penetrate the skin more easily. The facial pain caused by exposure to CX may cause the exposed soldier to remove a protective mask. Fortunately, a relatively high concentration of CX is required for toxic effects.
Mechanism of Injury. The mechanism of action of CX is not known. There are several theories for its action, including a necrotizing effect of the chlorine, the oxime, or the carbonyl group. The agent seems to cause the greatest systemic effects in the first capillary bed that is encountered. Cutaneous application or intravenous injection of CX causes pulmonary edema. Injection into the portal vein causes hepatic necrosis, but does not cause pulmonary edema.73
There are two main paths of injury:
- Direct: involving enzyme inactivation, corrosive injury, and cell death with rapid tissue destruction.
- Indirect: involving activation of alveolar macrophages, recruitment of neutrophils, and release of hydrogen peroxide, resulting in a delayed tissue injury such as the pulmonary edema.
The lethal dose in humans is not known, but is estimated to be about 30 mg/kg or a LCT50 of about 3200. The skin irritation is manifested at 1-3 mg/m3. The minimum effective respiratory dose is thought to be a CT of 300 mg/m3.
The German sources who investigated CX as a war gas felt that the respiratory toxicity was as impressive and possibly more dangerous than skin effects.49 They experimented with mixtures of Lewisite, mustard, and CX.
Clinical Effects. CX affects the skin, eyes, and lungs. The effects are quite rapid and phosgene oxime cause more severe tissue damage at a given concentration than other vesicant agents. The pain occurs immediately on contact with either the liquid or the solid form of CX.
A diagnostic characteristic of CX is this rapid onset of pain and irritation. Few other chemical agents produce such an immediate pain followed by rapid tissue necrosis. (Hydrofluoric acid and nitric acid are other such chemicals.)
Skin. CX is absorbed completely into the skin within seconds. CX is a powerful irritant that produces immediate effects varying from a mild irritation to severe local pain. When CX comes in contact with the skin, the site of contact becomes blanched within 30 seconds. The grayish blanched area is surrounded by erythema. The patient then may develop severe itching, hives, and painful blisters that resemble nettle stings. A wheal forms in about 30 minutes, and the blanched area turns brown in about 24 hours. A dark eschar forms during the next week. The eschar generally falls off in about three weeks, but some healing may be incomplete for as long as 4-6 months after exposure. Itching may be present throughout the healing.
The lesions of CX extend into the underlying panniculus and muscle. Congestion and hemorrhage are found with thrombosis in both small arteries and veins.
Eyes. The lesions caused by CX are similar to those caused by Lewisite. There is immediate pain and conjunctivitis. A Soviet scientist has stated that CX can cause blindness at low levels.49 There is no confirmation of this data by independent sources.
Airway. Inhaled CX causes rapid pulmonary edema. The pulmonary edema may be accompanied by a necrotizing bronchiolitis and thrombosis of the pulmonary vessels. Pulmonary edema also follows systemic absorption of large amounts of CX from the skin.There are no known long-term respiratory injury data.
Treatment. There is no antidote for CX, and there are no medications that are specific to the treatment of exposure to it. Analgesics are appropriate because of the intense pain caused by this substance.
Treatment of the skin lesions should be the same as for other causes of skin necrosis, with diligent attention to cleanliness and avoidance of secondary infection. The eye lesions should be treated as any other corrosive-substance eye injury.
Decontamination. Decontamination must be accomplished within seconds after contact to be effective. The military recommends use of the M291 decontamination kit with impregnated towelettes. They also recommend flushing the skin with large amounts of water as rapidly as possible to remove any CX that has not yet reacted with tissue. CX is readily soluble in water and is very soluble in organic solvents.
CX is considered a highly reactive agent and does not persist in the environment.
The M256A1 detector can detect CX and responds to levels within 3-5 mg/m3. It is possible to program the MM1 detector in the Fox vehicle to detect CX.
Methenamine pretreatment, used in U.S. Army munitions plants to protect workers from phosgene, may offer some protective effect against CX.73
Threat Analysis of the Vesicant Agents
Mustard is a chemical agent likely to be used on the modern battlefield and has been used by Iraq in very recent times. As noted in the covert production section, mustard is easy to make from commonly available materials with processes that are well described in the open literature. It should be presumed that terrorists may have access to small quantities of mustard gas and could employ it in confined spaces or at events with large attendance.
Mustard is quite attractive for use as a weapon of mass casualties because:
- It is fairly potent.
- It can inflict casualties despite appropriate use of respiratory protective devices.
- It is not easy to detect.
- It has delayed effects that allows the terrorist to escape the scene.
- It causes prolonged disability.
- It is stable in storage and can be transported easily.
- It is easy and inexpensive to produce.
- It is difficult to decontaminate.
- It is persistent in the environment.
- It penetrates many types of protective garments.
- It is deployed easily in explosive devices.
- It is effective in vapor, liquid, or aerosol forms.
Since mustard gas has a delayed presentation, many victims could be exposed before the agent is detected. This delay also means that the terrorist would be able to escape before the first casualties were detected.
The lack of an effective therapy and the heavily weighted casualty-to-lethality ratio would be significant pluses in the analysis of mustard as a terrorist’s weapon. There is a high likelihood of Mideast-trained terrorists having instructors who have had recent hands-on experience with mustard as participants in the Iran-Iraq War.
Mustard easily is delivered by rocket, bomb, aerosol spray tanks, liquid spray tanks, or explosive devices. Iraq used both aircraft and artillery to deliver mustard during the Iraq-Iran war.
The need for protective measures for handling and dispersing this agent is the major drawback for a terrorist’s use of mustard and analogues. The terrorist organization must accept casualties in the delivery team, use delayed fused munitions, or deliver the agent while dressed in protective gear. The first two scenarios are much more likely than the last. If the courier/delivery person is expendable, the potential delivery scenarios are mind-boggling.
Covert Production. Almost all producers of chemical weapons since World War I have manufactured the vesicant agents, principally sulfur mustard (bis[2-chloroethyl] sulfide). The chemical processes to produce these agents are well documented in the chemical literature and readily available from open sources. Indeed, the open literature includes data on reaction kinetics, catalysts needed, and appropriate operating parameters of the reactions. These chemicals can be made with primitive equipment if the producers are not overly concerned with worker health and safety or environmental impact.
There are nine separate chemical routes to this compound, none of which require sophisticated technology or special materials. At least three of these methods could be used by terrorists to produce small quantities of mustard gas. (Sulfur mustard easily can be prepared by bubbling ethylene through sulfur chloride or from hydrogen chloride and thiodiglycol.)
During World War I, thousands of tons of mustard gas were produced from alcohol, bleaching powder, and sodium sulfite. These are commonly available chemicals in high school chemistry laboratories.
More recent production involves chlorination of thiodiglycol, a common material with dual use as an ingredient in some plastics and ballpoint pen inks. Known as the Victor Meyer-Clarke process, the chlorination of thiodiglycol was developed by Germany during World War I and used extensively by Iraq in the 1980s. Drums of thiodiglycol, produced in the United States and diverted from the "intended" recipients were found by international inspectors in Iraq after the Gulf War. It is a one-step process and could be performed in a basement laboratory.
Processes for production of Lewisite and nitrogen mustards are equally simple. The major problem for the terrorist with covert production of mustard and similar agents would be casualties from accidental exposure to the agent.
References
1. Murphy S. Chemical warfare: The present position. Med War 1985; 1:31-39.
2. Grimaldi JV, Gugliotta G. Chemical plants feared as targets. The Washington Post. December 16, 2001:A01.
3. Weisse AB. From trench warfare to war on cancer. Hospital Practice 1992;30:141-148.
4. Jones FL. Chlorine poisoning from mixing household cleaners. JAMA 1972;222:1312.
5. Barrow CS, Alarie Y, Warrick JC, et al. Comparison of the sensory irritation response in mice to chlorine and hydrogen chloride. Arch Environ Health 1977;32:68-76.
6. Kaufman J, Burleons D. Clinical roentgenologic and physiologic effects of acute chlorine exposure. Arch Environ Health 1971;23: 39-44.
7. Hasan FM, Gehshan A, Fuleihan FJD. Resolution of pulmonary dysfunction following acute chlorine exposure. Arch Environ Health 2983;38:76-80.
8. Hedges JR, Morrisey WL. Acute chlorine gas exposure. JACEP 1979;8:59-63.
9. Sexton JD, Pronchik DJ. J Toxicol Clin Toxicol 1998;36:87.
10. Sciuto AM, Strickland PT, Kennedy TP, et al. Postexposure treatment with aminophylline protects against phosgene-induced acute lung injury. Exp Lung Res 1997;23:317-332.
11. Bosse GM. Nebulized sodium bicarbonate in the treatment of chlorine gas inhalation. Clin Toxicol 1994;32:233-241.
12. Douidar SM. Nebulized sodium bicarbonate in acute chlorine inhalation. Pediatr Emerg Care 1997;13:406-407. Accessed at NIH/NLM MEDLINE, HealthSTAR.
13. Vinsel PJ. Treatment of acute chlorine gas inhalation with nebulized sodium bicarbonate. J Emerg Med 1990;8:327-329.
14. Pascuzzi TA. Mass casualties from acute inhalation of chloramine gas. Mil Med 1998;163:102-104.
15. Fleta J, Calvo C, Zuniga J, et al. Intoxication of 76 children by chlorine gas. Human Toxicol 1986;5:99-100.
16. Chlorine: Guidelines in event of a deliberate release. Available at www.phls.org.uk/topics_az/deliberate_release/pdf/chlorine.pdf. Accessed 3-27-2003.
17. Richter W. Kampstoffwirkung und Heilung. Wehr und Wissenschaft. Vol. 26, 2nd ed. Leipzig, Gemany: Ambrosius Barth; 1941.
18. Pulmonary Agents. ccc.apgea.army.mil. Accessed 3-27-2003.
19. Frosolono MF, Pawlowski R. Effects of phosgene on rat lungs after single high level exposure. II. Ultrastructure changes. Arch Environ Health 1977;32:278-283.
20. Diller WF. Early diagnosis of phosgene overexposure. Toxicol Ind Health 1985;1:73-79.
21. World Health Organization, International Programme on Chemical Safety. Phosgene. Geneva: IPCS; 1997.
22. Diller WF. Medical phosgene problems and their possible solutions. J Occup Med 1975;32:271-277.
23. Fosolono MF, Pawlowski R. Effects of phosgene on rat lungs after single high level exposure. II. Ultrastructure changes. Arch Environ Health 1977;32:278-283.
24. Diller WF. Pathogenesis of phosgene poisoning. Toxicol Ind Health 1985;1:7-15.
25. Diller WF. Early diagnosis of phosgene overexposure. Toxicol Ind Health 1985;1:129-136.
26. Wells BA. Phosgene: A practitioner’s viewpoint. Toxicol Ind Health 1985;1:81-92.
27. Regan RA. Review of clinical experience in handling phosgene exposure cases. Toxicol Ind Health 1985;1:69-80.
28. Borak J, Diller WF. Phosgene exposure: Mechanisms of injury and treatment strategies. J Occ Environ Med 2001;43:110.
28. Sciuto AM, Stott RR, Hurt HH. Efficacy of ibuprofen and pentoxifylline in the treatment of phosgene-induced acute lung injury. J Appl Toxicol 1996;16:381.
29. Whitener DR, Whitener LM, Robertons KJ, et al. Pulmonary function measurements in patients with thermal injury and smoke inhalation. Am Rev Resp Disease 1980;122:731.
30. Lee CH, Guo YL, Tsai PJ, et al. Fatal acute pulmonary oedema after inhalation of fumes from polytetrafluoroethylene (PTFE). Euro Respir J 1997;10:1408-1411.
31. Stamgaugh JJ. Deadly toxin missing. Appalachian Focus, June 10, 2000. Accessed at http://www.knoxnews.com/news/10242.shmtl.
32. Urbanetti JS. Toxic inhalation injury. In: Medical Aspects of Chemical and Biological Warfare. Sidell F, Takafuji ET, Franz DR, eds. Walter Reed Army Medical Center, Washington, DC. Accessed at www.nbc-med.org/SiteContent/HomePage/WhatsNew/MedAspects/Ch-9electrv699.pdf. Accessed 3-31-2003.
33. Maidment MP, Rice P, Upshall DG. Retention of inhaled hexaflurocyclobutene in the rat. J Appl Toxicol 1994;14:395-400.
34. Harris DK. Polymer fume fever. Lancet 1951;II:1008-1011.
35. Robbins JJ, Ware RL. Pulmonary edema from Teflon fumes. N Engl J Med 1964;271:360-361.
36. Lewis CE, Kerby GR. An epidemic of polymer-fume fever. JAMA 1965:191:103-106.
37. Shusterman DJ. Polymer fume fever and other fluorocarbon pyrolysis related syndromes. Occ Med 1993;8:519-531.
38. Lailey AF. Oral N-acetylcysteine protects against perfluoroisobutene toxicity in rates. Human Exp Toxicol 1997;16:212-216.
39. Prakash UBS. Chemical warfare and bronchoscopy. Chest 1991; 100:1486-1487.
40. Frietag L, Firusian N, Stamatis G, et al. The role of bronchoscopy in pulmonary complications due to mustard gas inhalation. Chest 1991;100:1436-1441.
41. Report of the specialists appointed by the Secretary General to investigate allegations by the Islamic Republic of Iran concerning the use of chemical weapons. Document S/16433. New York: Security Council of the United Nations; 1986.
42. Borak J, Sidell FR. Agents of chemical warfare: Sulfur mustard. Ann Emerg Med 1992;21:303-308.
43. Eisenmenger W, Drasch G, von Clarmann M, et al. Clinical and morphological findings on mustard gas [Bis (2-Chloroethyl) Sulfide] Poisoning. J Forensic Sci 1991;36:1688-1698.
44. Carus WS. Chemical weapons in the Middle East. Washington, DC: The Washington Institute for Near East Policy; Research Memorandum 9. 1988.
45. Heully F, Gruninger M, et al. Collective intoxication caused by the explosion of a mustard gas shell. (Trans US Army) Annales d Medicine Legale 1956;36:195-204.
46. Papirmeister B, Gross CL, Meier HL, et al. Molecular basis for mustard-induced vesication. Fund Appl Toxicol 1985;5: S134-S149.
47. Smith KJ, Hurst CG, Moeller RB, et al. Sulfur mustard: Its continuing threat as a chemical warfare agent, the cutaneous lesions induced, progress in understanding its mechanism of action, its long term health effects, and new developments for protection and therapy. J Am Acad Derm 1995;32:765-776.
48. Papirmeister B, Feister AJ, Robinson SI, et al. Medical defense against mustard gas: Toxic mechanisms and pharmacological implications. Boca Raton, Fla.: CRC Press; 1991.
49. Rand Report on War Gas Exposure in the Gulf War. Skin Damaging Agents. Accessed at www.rand.org/publications/MR/MR1018.5/MR1018.5.chap3.html. Accessed 3-27-2003.
50. Pierard GE, Dowlati A, Dowlati Y, et al. Chemical warfare casualties and yperite-induced xerodermoid. American J Derm 1990; 112;565-570.
51. Requena L, Requena C, Sanches M, et al. Chemical Warfare: Cutaneous lesions from mustard gas. J Am Acad Dermatol 1988;19: 529-536.
52. Haber L. The Poisonous Cloud. Oxford: Clarendon Press; 1986.
53. Watson AP, Griffin GD. Toxicity of vesicant agents scheduled for destruction by the chemical stockpile disposal program. Environ Health Persp 1992;98:259-280.
54. Ruhl CM, Park SH, Danisa O, et al. A serious skin sulfur mustard burn from an artillery shell. J Emerg Med 1994;12:159-166.
55. Momeni AZ, Enshaaeih S, Meghdadi M, et al. Skin manifestations of mustard gas: A clinical study of 535 patients exposed to mustard gas. Arch Dermatol 1992:128:775-780.
56. Barranco V. Mustard gas and the dermatologist. Int J Dermatol 1991;30:684.
57. Momeni AZ; Aminjavaheri M Skin manifestations of mustard gas in a group of 14 children and teenagers: a clinical study. Int J Dermatol 1994;33:184-187.
58. Emad A, Rezaian GR. The diversity of the effects of sulfur mustard gas inhalation of respiratory system 10 years after a single heavy exposure. Chest 1997;112:734.
59. Beebe SW. Lung cancer in World War veterans: Possible relation to mustard-gas injury and 1918 influenza epidemic. JNCL 1960;25: 1231-1252.
60. Norman JE. Lung cancer mortality in World War I veterans with mustard-gas injury: 1919-1965. JNCI 1975;54:311-317.
61. Willems JL. Clinical management of mustard gas casualties. Ann Med Milit Belg 1989;3S:1-61.
62. Murray VSG, Volans GN. Management of injuries due to chemical weapons. BMJ 1991;302:129-130.
63. Smith KJ, Hurst CG, Moeller RB, et al. Sulfur mustard: Its continuing threat as a chemical warfare agent, the cutaneous lesions induced, progress in understanding its mechanism of action, its long term health effects, and new developments for protection and therapy. J Am Acad Derm 1995;32:765-776.
64. Yang YC, Baker JA, Ward JR. Decontamination of chemical warfare agents. Chem Rev 1992;92:1729-1743.
65. van Hooidonk C. CW agents and the skin: Penetration and decomposition. In: Proceedings of the International Symposium on Protection Against Chemical Warfare Agents, Stockholm, June 6-9, 1983:153-160.
66. Leonard RB, Teitelman U. Manmade disasters. Critical Care Clinics 1991;7:293-320.
67. Davis KG, Aspera G. Exposure to liquid sulfur mustard. Ann Emerg Med 2001;37:653-656.
68. Somani SM, Babu SR. Toxicodynamis of sulfur mustard. Int J Clin Pharm Therapy Toxicol 1989;27:419-435.
69. Eldad A; Ben Meir P, Breiterman S, et al. Superoxide dismutase (SOD) for mustard gas burns. Burns 1998;24:114-119.
70. Smith WJ, Dunn MA. Medical defense against blistering chemical warfare agents. Arch Dermatol 1991;127:1207.
71. Van Scott EJ, Kalmanson JD. Complete remission of mycosis fungoides lymphoma induced by topical nitrogen mustard (NH2). Cancer 1973;32:18-30.
72. AFMIC Special Weekly Wire 35-90A (U) Accessed at www.fas.org/irp/gulf/cia/970825/970613_ww35_90a_mic_txt_0001.html. Accessed 0-25-2001.
73. McAdams AJ Jr, Joffe MH. A toxico-pathologic study of phosgene oxime. Medical Laboratories Research Report 381. Army Chemical Center, MD; 1955.