Drug Criteria & Outcomes: Cinacalcet HCl (Sensipar) Formulary Evaluation
Drug Criteria & Outcomes: Cinacalcet HCl (Sensipar) Formulary Evaluation
Part 1 of 2: Indications, Mechanism of Action, Pharmacokinetics, Dosing & Administration, Adverse Events, amd Interactions
By April H. Eubanks,
PharmD Candidate
Harrison School of Pharmacy
Auburn (AL) University
Cinacalcet is the first in a class of calcimimetic agents that increases the sensitivity of the calcium-sensing receptor to activation by extracellular calcium.
Indications
Cinacalcet is indicated for the treatment of:
- Secondary hyperparathyroidism in patients with chronic kidney disease (CKD) on dialysis.
- Hypercalcemia in patients with parathyroid carcinoma.
Background on indications
Secondary hyperparathyroidism in patients with chronic kidney disease on dialysis:
Renal osteodystrophy, also known as renal bone disease, is a major cause of morbidity and mortality in patients with chronic kidney disease on dialysis. As renal failure progresses and glomerular filtration rate (GFR) declines, the kidneys’ ability to excrete phosphorus also declines, resulting in hyperphosphatemia. About 70% of all patients with end stage-renal disease (ESRD) have hyperphosphatemia, with approximately 40% having values that exceed 6.5 mg/dL (normal: 2.5-4.5 mg/dL).
High levels of phosphorus directly stimulate the release of parathyroid hormone (PTH). In addition, retention of phosphorus inhibits renal activation of vitamin D, which, in turn, reduces gut absorption of calcium. Low levels of calcium in the blood provide a major stimulus for secretion of PTH.
The parathyroid glands secrete PTH in an attempt to restore the normal balance of phosphorus and calcium. In normal patients, PTH release reduces reabsorption of phosphorus in the renal tubule and promotes its excretion. Thus, phosphorus and calcium levels return to normal. However, as functional renal mass declines, this renal response to PTH declines and hyperphosphatemia is not relieved. The resulting hypocalcemia causes PTH to stimulate the release of calcium through mobilization from bone.
Traditional agents used to treat hyperparathyroidism typically include vitamin D sterols and calcium-containing phosphate binders. These agents, however, put the patient at risk for developing hypercalcemia and may not be efficient at resolving the hyperparathyroidism.
Parathyroid carcinoma:
Hypercalcemia secondary to parathyroid carcinoma occurs as the result of uncontrolled release of PTH.
Mechanism of action
Cinacalcet prevents progression to renal osteo-dystrophy by decreasing PTH secretion. The calcium-sensing receptor located on the surface of the chief cell of the parathyroid gland is the principle regulator of PTH secretion. Cinacalcet increases the sensitivity of the calcium-sensing receptor to extracellular calcium, and thus, reduces the stimulation of PTH secretion. This reduction of PTH, in turn, reduces mobilization of bone.
Pharmacokinetics
Absorption and distribution
- After oral administration, maximum plasma concentration is achieved in approximately 2-6 hours.
- Cmax (maximum concentration) and AUC (area under the curve) were increased 82% and 68%, respectively, when cinacalcet was administered with a high-fat meal compared to fasting. Cmax and AUC were increased 65% and 50%, respectively, when cinacalcet was administered with a low-fat meal compared to fasting.
- Cinacalcet concentrations decline in a biphasic fashion with a terminal half-life of 30-40 hours.
- Steady-state is achieved within seven days.
- Volume of distribution is high (approximately 1,000 L), indicating extensive distribution.
- Cinacalcet is highly plasma-protein bound (93-97%).
Metabolism and excretion
- Primarily metabolized by CYP3A4, CYP2D6, and CYP1A2.
- In healthy volunteers,
a 75 mg dose of cinacalcet was rapidly and extensively metabolized via:
— oxidative N-dealkylation;
— oxidation of the naphthalene ring on the parent drug.
• Renal excretion is the primary route of elimination with approximately 80% of the dose recovered in the urine and 5% in the feces.
Recommended dosing and administration
- Cinacalcet should be taken with food or shortly after a meal.
- Cinacalcet tablets should not be divided. There have been no studies with the drug to suggest that it would be safe or that the drug would be efficacious when divided or crushed.
Secondary hyperparathyroidism in patients with CKD on dialysis
- Oral starting dose is 30 mg once daily.
- Serum calcium and phosphorus should be measured within one week.
- PTH should be measured one to four weeks after initiation or dose adjustment.
- Cinacalcet should be titrated no more frequently than every two to four weeks through sequential doses of 60, 90, 120, and 180 mg once daily to target PTH consistent with the NKF-K/ DOQI (National Kidney Foundation-Kidney Disease Outcomes Quality Initiative) recommendation for CKD patients on dialysis of 150-300 pg/mL.
- Cinacalcet may be used alone or in combination with vitamin D sterols and/or phosphate binders.
Parathyroid carcinoma
- Oral starting dose is 30 mg twice daily.
- The dose should be titrated every two to four weeks through sequential doses of 60 mg twice daily, 90 mg twice daily, and 90 mg three or four times daily as necessary to normalize serum calcium levels.
Special populations:
- Hepatic insufficiency: Patients with moderate-to-severe hepatic impairment (as indicated by the Child-Pugh method) should have PTH and serum calcium levels monitored closely throughout treatment due to higher AUC values (2.4-4.2 times higher) than seen in normal patients.
- Renal insufficiency: No dosage adjustments necessary.
- Geriatric patients: No dosage adjustments necessary.
- Pediatric patients: Not studied in patients younger than 18 years of age.
Monitoring
Serum calcium and phosphorus should be measured within one week, and PTH should be measured one to four weeks after initiation or dose adjustment. Once the maintenance dose has been established, serum calcium and phosphorus should be measured about every month, and PTH every one to three months.
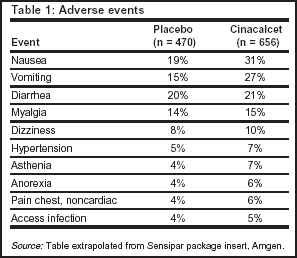
Adverse events
The incidence of serious adverse events was similar in the cinacalcet and placebo groups, 29% and 31%, respectively (see Table 1, below).
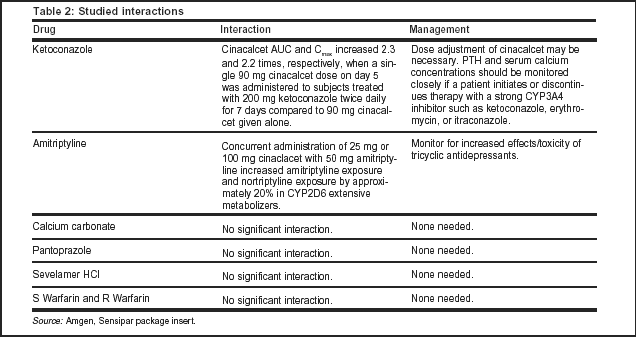
Drug interactions
Cinacalcet is a strong inhibitor of CYP2D6, but not CYP1A2, CYP2C9, CYP2C19, or CYP3A4. Cinacalcet also is a substrate of CYP1A2, CYP2D6, and CYP3A4 (see Table 2, below for more information on interactions).
Potential Interactions:
- CYP2D6 Substrates
Haloperidol Fluoxetine
Metoprolol Meperidine
Codeine Oxycodone
Paroxetine
In addition to these, any inducers or inhibitors of CYP1A2, CYP2D6, or CYP3A4 could potentially affect the metabolism of cinacalcet.
- Common Inducers:
Smoking Rifampin
Phenytoin Ritonavir
Phenobarbital Carbamazepine
- Common Inhibitors:
Ketoconazole Fluoxetine
Azole antifungals Grapefruit Juice
Cimetidine Macrolides
Cyclosporine Metronidazole
Amiodarone
Precautions
- Contraindication: Cinacalcet is contraindicated in patients with hypersensitivity to any component of the product.
- Seizures: Because seizure threshold is lowered significantly by reductions in serum calcium levels, patients, particularly those with history of a seizure disorder, should have serum calcium levels monitored closely during treatment.
- Hypocalcemia: Patients taking cinacalcet should be monitored closely for the occurrence of hypocalcemia. Potential manifestations include paresthesias, myalgias, cramping, tetany, and convulsions.
- Adynamic bone disease: If PTH levels are suppressed below 100 pg/mL (when assessed using the standard Nichols immunoradiometric assay), patients may develop adynamic bone disease.
- Hepatic insufficiency: Cinacalcet exposure as assessed by AUC in patients with moderate and severe hepatic impairment were 2.4 and 4.2 times higher, respectively, than that in patients with normal hepatic function. Patients with moderate and severe hepatic impairment should be monitored throughout treatment.
- Pregnancy Category C: No teratogenicity was observed in female rats when exposed during gestation to doses up to four times those of human exposure (180 mg/day) based on AUC comparison. Decreased fetal body weights were observed at all doses in conjunction with maternal decreased food intake and body weight gain.
Storage
Store at 25°C (77°F); when outside normal storage conditions, keep drug in temperature range of 15-30°C (59-86°F).
Part 1 of 2: Indications, Mechanism of Action, Pharmacokinetics, Dosing & Administration, Adverse Events, amd Interactions. Cinacalcet is the first in a class of calcimimetic agents that increases the sensitivity of the calcium-sensing receptor to activation by extracellular calcium.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.