Diabetic Ketoacidosis and Hyperosmolar Hyperglycemia — A Brief Review
Diabetic Ketoacidosis and Hyperosmolar Hyperglycemia A Brief Review
SPECIAL FEATURE
Financial Disclosure: Critical Care Alert's editor, David J. Pierson, MD, nurse planner Leslie A. Hoffman, PhD, RN, peer reviewer William Thompson, MD, executive editor Leslie Coplin, and managing editor Neill Kimball report no financial relationships relevant to this field of study.
INTRODUCTION
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) are two of the most common and serious acute complications of diabetes mellitus. DKA is responsible for more than 500,000 hospital days annually in the United States, at an estimated annual cost of $2.4 billion. Both conditions are part of the spectrum of uncontrolled hyperglycemia, and there is sometimes overlap between them. This article will discuss and compare the two conditions, with a focus on key clinical features, diagnosis, and treatment.
DIAGNOSTIC FEATURES
In DKA, there is an accumulation of ketoacids along with a high anion gap metabolic acidosis (see Table below).1 The acidosis usually evolves quickly over a 24-hour period. The pH is often < 7.20 and initial bicarbonate levels are often < 20 mEq/L. DKA patients (especially children) often present with nausea, vomiting, hyperventilation, and abdominal pain. Blood sugar levels in DKA tend to be 300-800 mg/dL, but they are sometimes much higher when patients present in a comatose state.
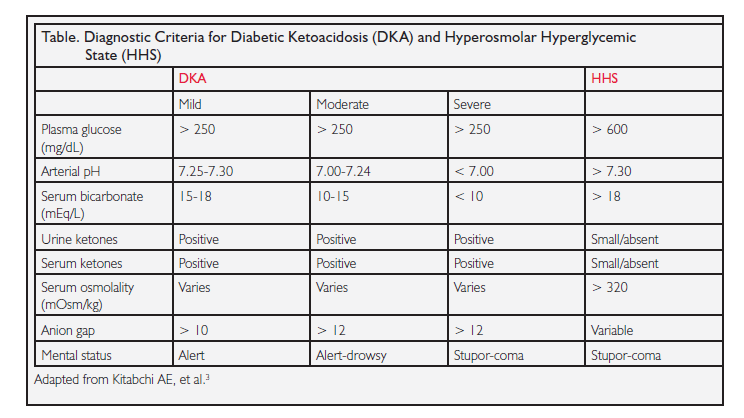
In HHS, there is no (or little) ketonemia but the plasma osmolality may reach 380 mOsm/kg, and as a result, patients often have neurologic complications such as coma. Bicarbonate levels are usually > 18 mEq/L. Blood sugars are more elevated in HHS, often > 800-1000 mg/dL. Neurologic symptoms are common in HHS. In some series, up to 50% of HHS patients present with coma. Abdominal pain is unusual in HHS.
Neurologic symptoms (obtundation and coma) are more common in HHS because hyperosmolality is more severe in HHS than in DKA. The hyperosmolality is due to the glucose osmotic diuresis causing water loss. However, severe acidosis in DKA can also cause significant neurologic deterioration. Neurologic symptoms typically occur when the effective plasma osmolality approaches 320 mOsm/kg. The effective osmolality is calculated by the equation:
[2 × Na (meq/L)] +
[glucose (mg/dL)/18].
Note that the effective plasma osmolality does not account for urea because it is a freely permeable molecule and its accumulation does not induce major intracellular osmotic changes in the brain. If the patient's serum osmolality has already been measured, the effective plasma osmolality can be calculated by this equation:
measured serum osmolality [BUN (mg/dL)/28].
Of note, stupor or coma in diabetic patients with an effective plasma osmolality < 320 mOsm/kg should prompt an investigation for another cause of the mental status change.
WHY ARE GLUCOSE LEVELS LOWER IN DKA?
DKA patients usually have lower blood sugar levels than HHS patients. One reason is that the acute acidosis in DKA causes distressing symptoms (e.g., nausea, dyspnea, abdominal pain) that encourage patients to seek attention at an earlier stage. In addition, DKA patients tend to be younger than HHS patients, and thereby have a higher glomerular filtration rate. Accordingly, DKA patients have a greater ability to excrete glucose in urine and can thereby limit the hyperglycemia. Of note, when end-stage renal patients develop severe hyperglycemia, they do not have the osmotic diuresis that leads to the extreme hyperosmolality. As a result, they rarely develop neurologic symptoms even though serum glucose levels may reach 1000-1500 mg/dL.
WHY DO PATIENTS HAVE ABDOMINAL PAIN?
DKA patients often have abdominal pain, but it is uncommon in HHS. The likely explanation for abdominal pain is ileus and delayed gastric emptying caused by the acidosis. In a prospective study of 200 hyperglycemic patients, abdominal pain occurred in 46% of DKA patients but in none of the HHS patients.2 The incidence of abdominal pain correlated with the severity of the acidosis. However, there was no relationship between abdominal pain and the degree of dehydration or the level of hyperglycemia. Of note, if abdominal pain is still present after the ketoacidosis resolves, other causes for the pain need to be considered.
INITIAL EVALUATION
When a patient presents with DKA/HHS, one should strive to identify the precipitant. The most common culprits are infection or inadequate insulin therapy (i.e., poor compliance). Various acute illnesses can trigger DKA/HHS, including stroke, myocardial infarction, and pancreatitis. Cocaine use can also trigger DKA. Some medications affect carbohydrate metabolism, including corticosteroids, thiazide diuretics, second-generation antipsychotics, and sympathomimetrics (e.g., dobutamine). New onset type 1 diabetics commonly present in DKA.
During initial investigation, one should consider other causes of metabolic acidosis and coma. If an anion gap acidosis is present, consider medications (e.g., metformin, aspirin), ingestions (e.g., methanol and ethylene glycol), sepsis/infection, and advanced chronic kidney disease. Note that none of these entities cause ketoacidosis. If a ketoacidosis is present, consider alcoholic ketoacidosis (AKA) and starvation ketosis. In an alcoholic presenting with ketoacidosis and normal blood sugars, AKA is almost always the diagnosis.
Physical examination will often show signs of volume depletion. DKA patients may have a fruity odor due to exhaled acetone. The hypovolemia causes peripheral vasoconstriction, so fever is rare, even when there is an infection.
Initial tests should include serum electrolytes (including anion gap), complete blood count with differential, urinalysis with ketones dipstick, serum ketones (if urine ketones are present), arterial blood gas (if urine ketones or anion gap are present), plasma osmolality, electrocardiogram, and chest X-ray. If indicated, additional tests might include cultures of blood, urine, and sputum; liver function tests; and lipase/amylase. Hemoglobin A1c may be useful to determine whether the patient's blood sugars have been poorly controlled prior to this episode.
Laboratory abnormalities in DKA depend on the patient's fluid intake, underlying renal function, osmotic diuresis, and level of insulin deficiency. The sine qua non of DKA is an elevated anion gap and metabolic acidosis. Most patients also have acute elevations in blood urea nitrogen (BUN) and creatinine due to hypovolemia. The severity of the anion gap acidosis depends on the rate of ketoacid production, duration of the episode, and rate of excretion in the urine. Thus, patients with normal renal function can minimize the anion gap by losing large quantities of ketoacids into urine.
Serum sodium levels in DKA/HHS depend on the balance between two forces the dilution of sodium caused by osmotic water movement out of cells and the concentration of sodium caused by glucosuria-induced water diuresis. Every patient is different, but in general, most patients with DKA/HHS present with mild hyponatremia. Rarely, some diabetics have such a brisk diuresis that they present with hypernatremia. When HHS patients have a normal or elevated serum sodium along with glucose levels > 1000 mg/dL, they are extremely hyperosmolar and at high risk for coma or seizure.
DKA/HHS patients have both potassium and phosphate deficits, even though most present with normal to high serum levels. The potassium deficit is a result of osmotic diuresis, insulin deficiency, and possibly gastrointestinal losses. Acidosis does not play a major role in the elevated serum potassium level seen in DKA. Hence, hyperkalemia also occurs in HHS. The phosphate deficit is a result of osmotic diuresis and decreased intake.
Most DKA/HHS patients present with a leukocytosis that is proportional to the ketonemia. Amylase and lipase are often elevated in DKA patients who do not have pancreatitis. Therefore, the diagnosis of pancreatitis in a DKA patient must be made using other clinical findings and radiology.
TREATMENT
Overall, the treatment principles for DKA and HHS are similar: intravenous fluids to restore volume, insulin to correct hyperglycemia, and electrolyte replacement. Protocols for DKA and HHS are readily available online and in the literature.1,3 The key points inherent to most of these protocols will be summarized.
Monitoring: Serum glucose should be measured hourly until stable. Serum electrolytes should be measured every 2-4 hours, depending on disease severity and response to therapy. In DKA, venous pH and serum beta-hydroxybutyrate measurements are an excellent way to monitor the response to therapy if your hospital can return blood chemistry results in a prompt manner. However, monitoring the anion gap is a simpler approach. Normalization of the anion gap indicates the ketoacidosis has corrected. Serial arterial blood gases are unnecessary because a venous pH is ~0.03 units lower than arterial pH and can provide enough data for decision making without the pain of an arterial stick.
Fluid replacement: The initial goals of fluid administration are expansion of intravascular volume and restoration of renal perfusion. Because of the osmotic diuresis, the average patient's volume deficit is 3-6 liters in DKA and 8-10 liters in HHS. The goal is to replace volume deficits within the first 24 hours. Fluid repletion is initiated with isotonic saline (0.9% NaCl) at a rate of 10-15 mL/kg/hr ideal body weight. This replaces the volume deficit, lowers plasma osmolality (since it is hypo-osmotic to the patient), and reduces serum glucose (both by dilution and by increasing renal perfusion), which in turn increases urinary glucose losses. Subsequent fluid selection will depend on serial assessments of electrolytes, urine output, and volume status. If the corrected sodium level is normal/high, patients are switched to 0.45% NaCl at 250-500 mL/hr. If the corrected sodium serum sodium level is still low, continue with 0.9% NaCl. Recall that hyperglycemia resolves more quickly than ketoacidosis (mean 6 hours vs 12 hours, respectively). Therefore, fluids are switched to 5% dextrose when the serum glucose reaches 200 mg/dL in DKA or 250-300 mg/dL in HHS. This allows continued insulin administration and prevents hypoglycemia, while waiting for the ketoacidosis to resolve.
Insulin: The mainstay of DKA/HHS treatment involves regular insulin, given via either continuous infusion or by frequent subcutaneous/intramuscular injections. Randomized trials have shown that insulin is effective regardless of the chosen route.4,5 Administering insulin through a continuous intravenous infusion allows more responsive control without the delayed onset and prolonged half-life issues of subcutaneous insulin. An initial bolus of insulin is not necessary provided the insulin is infused at a rate of 0.14 U/kg/hr (approximately 10 U/hour in a 70 kg patient).6 Dosing is the same in DKA and HHS. Regular insulin usually causes a fall in serum glucose by 50-70 mg/dL every hour. However, fluid repletion can also initially reduce serum glucose by 30-70 mg/dL per hour. Thus, the rate of fall may be more pronounced in HHS patients who are typically more volume depleted. The hyperglycemic episode is considered "resolved" when the anion gap has normalized, patients with HHS are mentally alert, the effective plasma osmolality is < 315 mOsm/kg, and the patient is able to eat. Patients with known diabetes who were previously treated with insulin may now be given insulin at their home dose. Insulin-naïve patients should be started on a multidose regimen (total 0.5-0.8 U/kg/day). The infusion should overlap the scheduled regimen by 1-2 hours to avoid rebound hyperglycemia.
Potassium: Despite a total body potassium deficit, the serum potassium concentration is often initially normal (or elevated). To prevent hypokalemia, KCl 20-30 mEq/L is added to the fluids once the serum potassium level falls < 5.3 mEq/L. If the patient is hemodynamically stable, 0.45% NaCl is preferred because adding potassium to 0.9% NaCl would result in a hypertonic solution (which would delay correction of hyperosmolality). The goal is to keep the serum potassium between 4-5 mEq/L. Since insulin will worsen hypokalemia, insulin therapy should be delayed until the serum potassium is > 3.3 mEq/L. Patients who present with hypokalemia must be aggressively repleted. Otherwise, insulin may cause possible arrhythmias, cardiac arrest, or respiratory muscle weakness.
Phosphate: DKA/HHS patients have a phosphate deficit that is unmasked by insulin initiation. However, the fall in serum phosphate during treatment is acute, self-limited, and rarely serious. Prospective randomized trials have not found that routinely replacing phosphate has any beneficial effect on duration of ketoacidosis, amount of insulin required, glucose control, morbidity, or mortality.7 In fact, it may have adverse effects such as hypocalcemia and hypomagnesemia. In general, phosphate replacement should only be given when serum levels are < 1.0 mg/dL or in patients with other indications such as cardiac dysfunction, respiratory depression, or hemolytic anemia.
Bicarbonate: Use of bicarbonate in DKA is controversial and there is no evidence of benefit.8 In a randomized trial of 21 DKA patients with an admission arterial pH of 6.90-7.14, bicarbonate therapy did not impact morbidity or mortality.9 No randomized trial has looked at patients with a pH < 6.90. There is theoretical benefit for patients with a pH < 7.00 and decreased cardiac contractility, but most experts agree it is not necessary if the pH is > 7.00.
IS ICU ADMISSION NECESSARY?
DKA patients are often admitted to an ICU even though there is a low risk of mortality in this condition. In a recent observational study of 159 acute care hospitals in New York state, Gershengorn et al found significant variation in ICU admission practices for DKA patients.10 Of the 15,994 patients studied, 53% were admitted to an ICU. Patients were more likely to be admitted to the ICU if they lived in a more affluent zip code, had more chronic illnesses, were admitted after an emergent presentation to the hospital, or were admitted on the weekend. ICU admission was less likely for older and nonwhite patients. ICU admission was more likely if the hospital had a higher utilization of ICU level care for non-DKA diagnoses. ICU admission was less likely if the hospital had a high volume of DKA patients. There was no association between ICU admission and hospital length of stay or mortality. In a multilevel regression model, more than half of the variation in ICU admission practice attributable to hospitals remained unexplained. Overall, this study suggests that many DKA patients can probably be safely managed outside the ICU. However, hospital staffing and policies will need to be aligned with the nursing demands and close monitoring inherent to DKA management.
REFERENCES
1. Kitabchi AE, et al. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2009;32:1335-1343.
2. Umpierrez G, Freire AX. Abdominal pain in patients with hyperglycemic crises. J Crit Care 2002;17:63-67.
3. Kitabchi AE, et al. Thirty years of personal experience in hyperglycemic crises: Diabetic ketoacidosis and hyperglycemic hyperosmolar state. J Clin Endocrinol Metab 2008;93:1541-1552.
4. Umpierrez GE, et al. Efficacy of subcutaneous insulin lispro versus continuous intravenous regular insulin for the treatment of patients with diabetic ketoacidosis. Am J Med 2004;117:291-296.
5. Fisher JN, et al. Diabetic ketoacidosis: Low-dose insulin therapy by various routes. N Engl J Med 1977;297:238-241.
6. Kitabchi AE, et al. Is a priming dose of insulin necessary in a low-dose insulin protocol for the treatment of diabetic ketoacidosis? Diabetes Care 2008;31:2081-2085.
7. Wilson HK, et al. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med 1982;142:517-520.
8. Viallon A, et al. Does bicarbonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med 1999;27:2690-2693.
9. Morris LR, et al. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med 1986;105:836-840.
10. Gershengorn HB, et al. Variation in use of intensive care for adults with diabetic ketoacidosis. Crit Care Med 2012;40:2009-2015.
Adapted from Kitabchi AE, et al.3
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.