Cardiac Disorders in the Pediatric Patient
Authors: Todd W. Wylie, MD, Assistant Clinical Professor, University of Florida, Shands Healthcare, Jacksonville; Ghazala Q. Sharieff, MD, FACEP, FAAEM, FAAP, Associate Clinical Professor, Children’s Hospital and Health Center/University of California, San Diego; Director of Pediatric Emergency Medicine, Palomar-Pomerado Hospital/California Emergency Physicians, San Diego, CA.
Peer Reviewer: William J. Brady, MD, Associate Professor of Emergency Medicine and Internal Medicine and Vice Chair, Emergency Medicine, University of Virginia, Charlottesville.
Although pediatric cardiac diseases infrequently are seen in the emergency department (ED), early diagnosis and aggressive management is critical. Most importantly, the clinician must include these diseases in their differential and have a thorough understanding of typical and atypical presentations for congenital heart disease (CHD), dysrhythmias, myocarditis and pericarditis. Any child who has a clinical presentation suggestive of cardiac disease, must receive appropriate diagnostic testing and timely referral to optimize the child’s outcome. The authors provide a thorough, focused review of the most commonly encountered cardiac diseases in the ED and key aspects to stabilization. — The Editor
Introduction
Pediatric cardiac disorders, though not common in the ED, may be associated with significant morbidity and mortality. Emergency physicians cannot afford to miss these problems; a missed diagnosis may result in further decompensation or death. Further challenging the clinician, the presenting complaints for many pediatric cardiac disorders may be nonspecific. Vague chief complaints such as fussiness or difficulty feeding may be the initial presentation for an infant with supraventricular tachycardia or congestive heart failure (CHF). Complaints commonly associated with cardiovascular disorders in adults, such as chest pain or palpitations, may be absent in children. This article provides an overview of the diagnosis and management of pediatric cardiac disorders.
Dysrhythmias
Normally, the heart rate is fastest in the newborn, and decreases with age until adolescence, when the upper and lower parameters of normal are similar to those of an adult. Normal variations do occur, but the explanation should follow expected physiologic patterns. Sinus tachycardia may result from fever, pain, anxiety, dehydration, anemia, or acute blood loss. Sinus bradycardia may be caused by vagal stimulation, hypoxemia, increased intracranial pressure, hypothyroidism, or acidosis. A sinus dysrhythmia is not uncommon in children, and does not necessarily denote a pathological condition.
Supraventricular Tachycardia. Paroxysmal supraventricular tachycardia (SVT) is the most common symptomatic dysrhythmia in infants and children. (See Figure 1.) The history for infants with SVT may include nonspecific complaints, such as fussiness, lethargy, poor feeding, pallor, sweating with feeds, or simply "not acting right." Chest pain, pounding in the chest, dizziness, or shortness of breath may be the initial complaint of older children with SVT. In contrast, patients with sinus tachycardia generally have a more specific history, such as fever, dehydration due to fluid or blood loss, anxiety, or pain that accounts for the tachycardia.
Figure 1. Rhythm Strip Demonstrating Supraventricular Tachycardia
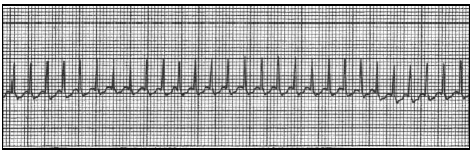
In newborns and infants with SVT, the heart rate is greater than 220 beats per minute (BPM).1 SVT in older children is defined as a heart rate of more than 180 BPM.1 The electrocardiogram (ECG) for supraventricular tachycardia will show a narrow complex tachycardia without discernible p-waves or beat-to-beat variability. (See Figure 1).
An accessory atrioventricular pathway is the most common cause of SVT in children younger than 12 years, whereas atrioventricular node re-entry tachycardia becomes more common in adolescents.2 The term Wolff-Parkinson-White (WPW) syndrome is applied when there is an extra atrioventricular accessory pathway.
Treatment for supraventricular tachycardia depends on patient stability. Immediate cardioversion with 0.5 to 1 J/kg is indicated for unstable patients. In the stable patient, vagal maneuvers, such as blowing through an occluded straw or placing an ice bag on an infant’s face, may prove successful. If vagal maneuvers are unsuccessful, adenosine 0.1 mg/kg (up to 6 mg) may be given through the most central vein possible. A normal saline flush of 5 mL should then be administered. If the first dose is unsuccessful, an increased dose of 0.2 mg/kg (up to 12 mg) may be administered.1 If these measures are unsuccessful, a pediatric cardiologist should be consulted; there are several options, including procainamide or amiodarone. Verapamil should be avoided in children younger than 1 year; cardiovascular collapse and death can occur due to electromechanical dissociation.3 Long-term management of children with supraventricular tachycardia may include beta-blockers, procainamide, sotalol, amiodarone, or flecainide. Another option may be radio-frequency catheter ablation, which has reported success rates of 85-95%.4
Digoxin is effective in converting SVT; however, the onset of action is slow, and it may be associated with dangerous complications if used to treat a patient with SVT and WPW.5
Wolff-Parkinson-White Syndrome (WPW). WPW syndrome denotes an accessory atrioventricular pathway (Kent bundle) that predisposes individuals to supraventricular tachycardias. Episodes of SVT in children with WPW syndrome usually occur early in the first year of life, frequently resolve, and then recur later in life, usually between 6-8 years of age.6
Clinical manifestations of WPW syndrome are secondary to the occurrence of dysrhythmias and dependent on the patient’s age, and the duration and rate of the dysrhythmia. Infants typically present with symptoms of fussiness, irritability, and poor feeding. If CHF is present, caretakers may describe pallor, cough, and respiratory distress. The heart rate in infants will be greater than 220 BPM, and may be as fast as 280 BPM.7 The presentation in older children differs due to their ability to describe symptoms and the fact that CHF rarely develops secondary to SVT in older children.7 Episodic palpitations are a common complaint in older children, and often occur during rest. Older patients also describe symptoms of dizziness, shortness of breath, and chest discomfort.
SVT in WPW syndrome generally is initiated by a premature atrial depolarization that travels to the ventricles via the normal atrioventricular pathway, travels retrograde through the accessory pathway and reenters the AV node to start a reentrant type of tachycardia.8 Antegrade conduction through the AV node followed by retrograde conduction through the accessory pathway produces a narrow complex tachycardia (orthodromic tachycardia), and is the most common form of SVT found with WPW syndrome.5 Less commonly, reentry occurs with antegrade conduction through the accessory pathway and retrograde conduction through the AV node (antidromic tachycardia), to produce a wide complex tachycardia.5
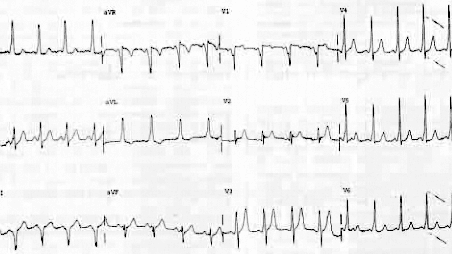
ECG findings consistent with WPW syndrome include a short PR interval, a widened QRS, and a slurred upstroke of the QRS complex known as a delta wave. (See Figure 2.) The characteristic ECG findings of WPW syndrome are not evident during a tachydysrhythmia. As described earlier, the ECG will demonstrate a narrow QRS supraventricular tachycardia if conduction is antegrade through the AV node (orthodromic). A wide QRS tachycardia may be present with antegrade conduction through the accessory pathway (antidromic), and this can be mistaken for a ventricular tachycardia. Atrial fibrillation in patients with WPW syndrome, although rare in infancy, does occur in late childhood, particularly after 12 years of age.7
Figure 2. ECG Showing Wolff-Parkinson-White (WPW)
Syndrome
Note the delta waves.
Acute management of SVT in patients with WPW syndrome is similar to that of other forms of SVT with some specific precautions. In unstable patients, synchronized cardioversion at 0.5 to 1 J/kg is the recommended initial therapy.1 Repeat attempts at cardioversion with 2 J/kg are acceptable if the initial attempt is unsuccessful.1 If intravenous access is available immediately, adenosine at a dose of 0.1 mg/kg as a rapid IV bolus (maximum of 6 mg for initial dose) is also acceptable as initial therapy.1 Subsequent doses of adenosine can be doubled to 0.2 mg/kg and again to 0.4 mg/kg (maximum of 12 mg) as needed.9 Cardioversion should not be delayed for attempts at intravenous access or sedation in unstable patients. In stable patients, adenosine (same dose as above) is the first line of therapy. Failure to terminate the dysrhythmia with adenosine in a stable patient should prompt consultation with a pediatric cardiologist. Digoxin should not be used in patients with WPW syndrome secondary to the risk of enhancing conductivity through the accessory path and subsequent ventricular fibrillation and sudden death. It also should be noted that intravenous verapamil and propranolol are contraindicated in children younger than 1 year of age.7 Following stabilization and conversion of the SVT to a normal sinus rhythm, long-term prophylactic therapy should be instituted by a pediatric cardiologist. Generally, prophylactic therapy is instituted in all infants until at least 1 year of age.5 Beta-blockers are recommended as the drug of choice for prophylactic therapy in infants with WPW syndrome.5 Procainamide may be used if beta-blockers fail.5
Bradydysrhythmias. Acute bradycardia, in pediatric patients, may be attributable to vagal stimulation, hypoxemia, acidosis, or increased intracranial pressure. Complete heart block is a common cause of significant bradycardia in pediatric patients and may be acquired or congenital. Causes of congenital heart block include structural lesions like L-transposition of the great arteries, or maternal connective tissue disorders. Acquired heart block may result from disorders such as Lyme disease, lupus, muscular dystrophies, Kawasaki disease, or rheumatic fever.8,10
Management of bradycardia includes identification of the etiology and appropriate cardiopulmonary resuscitation, with assisted ventilation, oxygenation, and chest compressions as indicated. Epinephrine or atropine is given if symptomatic bradycardia persists despite initial resuscitative measures.1 Pharmacologic management of complete heart block includes atropine or isoproterenol. A transcutaneous pacemaker may serve as a bridge to a transvenous pacemaker in the acute setting.
Ventricular Tachycardia/Fibrillation. Ventricular tachycardia (VT) may result from electrolyte abnormalities, congenital heart disorders, myocarditis, or drug toxicity. Both ventricular tachycardia and ventricular fibrillation (VF) are uncommon rhythms in pediatric patients. VT with a pulse in an unstable patient warrants immediate synchronized cardioversion at 0.5 to 1 J/kg.1 Pharmacologic alternatives also may include amiodarone, procainamide, or lidocaine.1 Amiodarone and procainamide should not be given together. Pulseless VT and VF should be treated with defibrillation at 2 J/kg, then 2-4 J/kg, and then 4 J/kg if it does not respond to the initial attempts at defibrillation.1 If defibrillation is unsuccessful, epinephrine should be given, and repeated every 3-5 minutes as necessary.1 Amiodarone, lidocaine, and magnesium also may be considered if VF or pulseless VT is refractory to the above measures.1
Prolonged QT Syndrome. Congenital prolonged QT syndrome, also known as long QT syndrome (LQTS), is a disorder of delayed ventricular repolarization characterized by prolongation of the QT interval. (See Figure 3.) It may be either hereditary or acquired. Jervell-Lange-Nielsen syndrome is an autosomal recessive form of prolonged QT syndrome associated with congenital deafness, whereas Romano-Ward syndrome is an autosomal dominant form that is not associated with deafness. The hallmark dysrhythmia is torsades de pointes (twisting of the points), although other dysrhythmias may occur. Patients commonly present between the ages of 9 and 15 years with recurrent episodes of near-syncope or syncope.11,12 Syncopal episodes may be precipitated by intense emotion, vigorous physical activity, or loud noises. Spontaneous return of consciousness usually follows a syncopal episode, but the dysrhythmia has the potential to degenerate into ventricular fibrillation and sudden death.11,13 Approximately 10% of children with LQTS present with sudden death, with younger children being more likely to die suddenly.11-13 Several reports of death due to abrupt awakening by an alarm clock or ringing telephone have been documented in pa-tients with LQTS.11,13 LQTS also may present in infancy as sudden infant death syndrome.14-16
The Bazett formula is the most commonly used manual method to measure the QT interval. (See Figure 3.)17 This equation accounts for the normal physiologic shortening of the QT interval that occurs with increasing heart rate. The QT interval on ECG represents the time from onset of depolarization to completion of repolarization. Lead II generally is the lead accepted for QTc calculations. The QT interval is measured from the onset of the QRS complex to the end of the T wave where it returns to the baseline. The QT and preceding RR intervals should be measured for three consecutive beats and averaged for the greatest accuracy.18 Other ECG abnormalities that suggest LQTS include prominent U waves, a T-U complex with an indistinct termination of the T wave, and broad T waves, which may be notched, biphasic, or inverted. T-wave alternans, an alternating amplitude and polarity of the T waves, also may be present.19
Computer-generated interpretations of ECGs generally are useful and correct, but may be less accurate as a screening tool for LQTS.20 Errors in the automated measurement of the QTc increase when the precise end of the T wave cannot be determined easily. One recent study shows that, despite a prolonged QTc by automated measurement, the computer generated a normal ECG reading in 50% of family members who proved to be genetic carriers of LQTS.20
Any patient with a suspicious history, borderline prolongation of the QT interval with symptoms, or identified prolonged QT syndrome should receive a cardiology consult for further management. Therapy is aimed at reducing sympathetic activity to the heart, either pharmacologically or surgically. Beta-blockers generally are recommended as the initial therapy of choice and have been shown to significantly reduce episodes of syncope and sudden death, with a decrease in mortality from 71% in untreated patients to 6% in those treated.21 Beta-blockers also have been shown to effectively eradicate dysrhythmias in 60% of patients.12 These beneficial effects occur, however, in the absence of QT shortening.13 All beta-blockers appear to be effective, but propranolol and nadolol are used most commonly.22
Patients presenting with LQTS may require emergency intervention. Patients with polymorphic ventricular tachycardia or torsades de pointes of unknown etiology, should receive IV magnesium (25-50 mg/kg; max 2 g). Serum electrolytes and a toxicology screen should be obtained. Beta-blockers may be useful in suppressing catecholamine surges and further dysrhythmic activity. Patients with recurrent ventricular tachycardia may require temporary transcutaneous ventricular pacing.23
Physicians should be aware of drugs known to prolong the QT interval and avoid their use in patients with LQTS. Family members and close friends should be instructed in cardiopulmnary resuscitation because of the high risk of unexpected cardiac events. Once a patient is diagnosed with LQTS, ECGs should be done on all other family members. Asymptomatic carriers often are identified by routine ECG or after familial ECG screening due to a symptomatic family member.
Brugada Syndrome. First described in 1992 by Brugada and Brugada, the Brugada syndrome is associated with sudden death in patients with a structurally normal heart, and characterized by specific ECG findings, which include a right bundle-branch block (RBBB) and ST-segment elevation in leads V1-V3.24 Although it has been found throughout the world, occurring in both women and children,24-28 the syndrome most commonly occurs in southeast Asian males, and the initial cardiac event usually occurs in the 30s and 40s.25,29,30 It also has been suggested that Brugada syndrome may cause sudden death in children during the first few months of life and may be misdiagnosed as sudden infant death syndrome.27
Brugada syndrome is a genetically inherited disease with an autosomal dominant inheritance pattern and variable expression.25,27,30 The mutation occurs in the SCN5A gene that encodes for the human cardiac sodium channel. The improperly functioning sodium channels predispose to developing ventricular tachydysrhythmias, specifically a rapid polymorphic ventricular tachycardia, which can degenerate into ventricular fibrillation.26,30 Dysrhythmic episodes are unpredictable and may or may not terminate spontaneously. Episodes that do not terminate spontaneously result in sudden cardiac arrest, whereas those that do terminate spontaneously may present as syncope or near-syncope. Unfortunately, sudden death may be the initial presentation. Sudden death seems to occur commonly during sleep, particularly in the early hours of the morning.30 Medications, such as Type I antidysrhythmics, which affect the cardiac sodium channels, can provoke dysrhythmias.26
A presumptive diagnosis of Brugada syndrome should be made in patients with sudden cardiac arrest that are resuscitated successfully, syncope, or near-syncope, and have an ECG pattern suggestive for Brugada syndrome. The ECG pattern, as originally described, consists of a RBBB with ST-segment elevation in leads V1-V3.24 It has been noted since that the RBBB pattern may be incomplete.26 Three types of ST-segment patterns have been described. Type 1 is characterized by a coved ST-segment elevation with amplitude of more than 2 mm at its peak followed by a negative T-wave.30 The ST-segment pattern in Type 2 has a high take-off ST-segment elevation that descends, followed by a positive T-wave resulting in a saddle-back configuration.30 Type 3 has a ST-segment elevation of less than 1 mm with a saddle-back or coved morphology.30 The morphology of the ST-segment elevation does not appear to be predictive of sudden cardiac events.29 It is important to note that the ECG pattern is intermittent and may transiently normalize.29,30
Management in the ED of symptomatic patients with a suggestive ECG includes initial stabilization as necessary and cardiology consultation. All patients suspected of having Brugada syndrome require confirmatory testing and definitive management by a cardiologist. The only effective long-term treatment for patients at risk for sudden cardiac events is placement of an implantable cardioverter-defibrillator (ICD). Antidysrhythmic medications have not been shown to have any beneficial effects in patients with Brugada syndrome.
Further inpatient studies, such as administration of pharmacologic agents to accentuate the ECG changes or electrophysiologic studies, can be performed to confirm the diagnosis of Brugada syndrome. Although there has been some debate, these studies appear to be useful as predictors of dysrhythmic events.31,32
Congenital Heart Disease (CHD)
CHD occurs in eight of 1000 live births; many of the structural CHDs present in the neonatal period.33 The signs and symptoms of CHDs may be nonspecific and include: tachypnea, sudden onset of cyanosis or pallor that worsens with crying, sweating with feedings, lethargy, or failure to thrive. Time of presentation for cyanotic and acyanotic CHD and the common associated ECG and pulmonary blood flow pattern findings are listed in Tables 1 and 2.34
Table 1. Typical ECG and Radiographic Findings in Cyanotic
Congenital Heart Disease Based on Age of Presentation

Table 2. Age of Presentation, ECG Findings,
and Pulmonary
Blood Flow Patterns with Acyanotic
Congenital Heart Disease
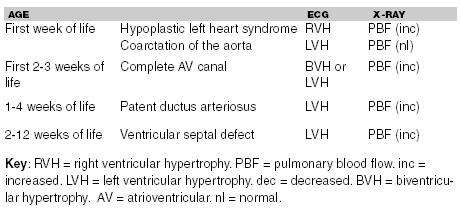
Congenital heart lesions that present in the first two to three weeks of life are typically ductal-dependent. The ductus arteriosus sustains blood flow for these infants and when the ductus closes anatomically at 2-3 weeks of life, these infants decompensate. The ductus arteriosus supplies blood to either the lungs, as with tetralogy of Fallot (TOF) or tricuspid atresia (cyanotic CHD), or to the systemic circulation as in the case of coarctation of the aorta, hypoplastic left heart syndrome, or aortic stenosis. When the ductus closes, the infant develops respiratory distress, shock, or an altered mental status.
Any neonate presenting with cyanosis or respiratory distress should be evaluated immediately with close attention to the ABCs (airway, breathing, and circulation). A pulse oximetry measurement or arterial blood gases (ABG) measurement from the right hand and foot may help in the diagnosis. Significantly lower oxygen saturation will be found in the lower extremities in a patient with a coarctation of the aorta or interrupted aortic arch. However, transposition of the great arteries may be associated with higher oxygen saturations in the lower extremities than in the upper extremities.35
The main etiologies of cyanotic CHD are TOF, tricuspid atresia, transposition of the great arteries (TGA), truncus arteriosus, total anomalous pulmonary venous return (TAPVR), and pulmonary atresia or stenosis. If cyanosis is present, one must first determine if the cyanosis is central (cyanosis of the lips, tongue, and oral mucous membranes) or peripheral. If the cyanosis is central, then performing the hyperoxia test can help distinguish a cardiac from a pulmonary etiology. To perform the hyperoxia test, the cyanotic infant should be placed on 100% oxygen. If the oxygen saturation increases significantly, then the infant probably has pulmonary pathology. If the oxygen saturation does not increase by 10%, then consider a congenital heart disorder. The most accurate way to perform this test is to obtain a baseline ABG measurement, and then repeat it approximately 10 minutes after administration of 100% oxygen.
Other congenital cardiac lesions that present in the first month of life are the left-to-right intracardiac shunts, such as ventricular septal or atrioventricular canal defects. As the normal pulmonary vascular resistance falls during the first month of life, any pre-existing left-to-right shunt will see a gradual increase in flow across the shunt resulting in CHF. The differential diagnosis of congenital heart diseases that cause CHF include the left-to-right intracardiac shunts, hypoplastic left ventricle, coarctation of the aorta, truncus arteriosus, endocardial cushion defect, patent ductus arteriosus (PDA), aortic stenosis, interrupted aortic arch, aortic atresia, and mitral valve atresia.36,37
An ECG and chest X-ray should be obtained in all infants suspected of having CHD. Frequently it is difficult in the ED to distinguish between sepsis and CHD. Therefore, all children who present with cyanosis or shock should receive a full septic evaluation and early initiation of antibiotic therapy. The lumbar puncture may be postponed if there is any concern about the infant’s respiratory status.
If a ductal-dependent CHD is suspected, a prostaglandin E1 (PGE1) infusion should be initiated immediately at a rate of 0.05-0.1 mcg/kg/minute. Ideally, a pediatric cardiologist should be consulted prior to initiation of the infusion and echocardiography used if immediately available to confirm the diagnosis of CHD.38 However, therapy with PGE1 should not be withheld; there are no other temporizing medications to maintain ductal patency. Prostaglandin is a very potent vasodilator and will have immediate effects on the ductus, with improvement usually seen within 15 minutes. The practitioner should be prepared to intubate because apnea is a common occurrence with PGE1 initiation. Although routine intubation is not necessary prior to initiation of PGE1, it may be prudent to prophylactically intubate patients requiring transportation to another facility. Other complications of prostaglandin use include fever, hypotension, and seizures.
If the patient has CHF, furosemide (1 mg/kg) should be administered with the possible addition of morphine, dobutamine, dopamine, or angiotensin inhibitors.39 These patients can deteriorate rapidly and should be admitted to either a neonatal or pediatric intensive care unit (PICU).
Hypertrophic Cardiomyopathy
Most cases of hypertrophic cardiomyopathy (HCM) are diagnosed in patients between 30 and 40 years of age, but 2% of cases occur in children younger than 5 years, and 7% occur in children younger than 10 years of age.40 The hallmark anatomic finding in patients with HCM is an asymmetric, hypertrophied, nondilated left ventricle with greater involvement of the septum than the ventricle. Although the left and right ventricles are small to normal in size, there is atrial enlargement and thickening of the mitral valve in 95% of cases.41
Presenting symptoms range from chest pain, palpitations, or shortness of breath, to near syncope, syncopal episodes, and sudden death. Children with HCM who experience syncope are at significant risk for sudden death, typically occurring during strenuous exercise.
The physical examination typically reveals a loud S4 gallop with a harsh mid-systolic crescendo-decrescendo murmur. This murmur is accentuated with Valsalva maneuvers or the standing position. Squatting, isometric hand grip, or lying down will decrease the murmur due to an increase in left ventricular end-diastolic volume.
Classic ECG findings include left atrial and ventricular hypertrophy, ST-segment abnormalities, T-wave inversions, Q waves, and diminished or absent R waves in the lateral leads. Premature atrial and ventricular contractions, supraventricular tachycardia, multifocal ventricular dysrhythmias, or new onset atrial fibrillation also may be present. The echocardiogram is the diagnostic procedure of choice.
A cardiologist should be consulted as soon as the diagnosis is suspected to assist with management. Unless the patient is symptomatic, it is not necessary to start medications in the ED. The goal of therapy is to decrease the heart rate to increase the diastolic filling time with beta-blockers the cornerstone of therapy. Calcium-channel blockers also are used, particularly if there is no response to the beta-blocker. Nitrates should be avoided if HCM is suspected, as they decrease ventricular volume and resultant outflow tract volume. Digoxin also is contraindicated.
Ventricular dysrhythmias may be treated with amiodarone. Surgical intervention, typically a septal myomectomy, may be warranted in patients with systolic gradients more than 50 mmHg, minimal response to medical management, or severe symptoms. Antibiotic prophylaxis against bacterial endocarditis also is indicated. Patients undergoing an evaluation for HCM, or in whom the diagnosis is suspected, should refrain from strenuous activity or physical exertion as sudden death can occur.42
Myocarditis
Myocarditis is an inflammatory condition of the myocardium that can occur in conjunction with pericarditis or in isolation. Although there are numerous causes, including infections, drugs, endocrine disorders, radiation, and collagen vascular diseases, the most common etiology in North America is viral (coxsackievirus A and B, ECHO viruses, and influenza viruses).40,43 The clinical presentation varies depending on multiple factors, including the etiology and age of the patient. Neonates and infants may present with symptoms such as lethargy, poor feeding, irritability, pallor, fever, or failure to thrive. Symptoms suggestive of heart failure, such as diaphoresis with feeding, rapid breathing, or respiratory distress, may be present. Older children and adolescents can present similarly, and also have complaints of weakness, fatigue, chest pain, or shortness of breath. A recent history of a nonspecific viral-syndrome type illness also is common. Potential findings on physical examination include tachypnea, tachycardia, hyperthermia or hypothermia, and hypotension. Signs of poor perfusion and heart failure, such as tachycardia, weak pulses, decreased capillary refill, cool mottled extremities, jugular venous distention, hepatomegaly, and lower extremity edema, may be present. Heart tones may include an S3, that may be muffled if pericarditis is present. Although several types of dysrhythmias occur, the most common dysrhythmia is sinus tachycardia. Tachycardia faster than expected for the degree of fever (10 BPM for each degree of temperature elevation) may be suggestive of myocarditis.
A complete blood count (CBC) with differential may show an elevated white blood cell count. A lymphocytic predominance on the differential suggests a viral etiology. The sedimentation rate and C-reactive protein level usually are elevated, but normal values do not exclude the diagnosis of myocarditis. Elevation of creatine kinase-MB isoenzyme (CK-MB), lactate dehydrogenase (LDH), and troponin levels can occur. Blood cultures as well as throat, nasopharyngeal, stool, and urine specimens should be obtained to identify bacterial or viral pathogens.
The most common finding on ECG is sinus tachycardia. Other abnormalities, such as premature ventricular beats, junctional tachycardias, ventricular tachycardias, and even second- and third-degree atrioventricular blocks also may be present. A low-voltage QRS, fewer than 5 mm in all limb leads, suggests myocarditis. As pericarditis may occur simultaneously, the ECG may show ST-segment elevation and PR depression (see section on pericarditis).
The chest x-ray frequently demonstrates cardiomegaly, and pulmonary edema may be present. An echocardiogram is valuable to determine myocardial function. If heart failure is present, an echocardiogram will show increased left ventricular end diastolic and systolic dimensions. Other potential echocardiogram findings include left ventricular wall dysfunction, decreased ejection fraction, segmental wall motion abnormalities, or global hypokinesis.
Initial management should focus on the patient’s respiratory and circulatory status. All patients in respiratory distress should be started on supplemental oxygen, and cardiac monitoring and pulse oximetry should be initiated. If the patient continues to deteriorate, or is in cardiogenic shock, endotracheal intubation and ventilatory support may be necessary. Management for patients with signs of heart failure includes diuretics, digoxin, inotropic agents as needed, and afterload reduction. Intravenous furosemide (1 mg/kg IV) is used to decrease fluid overload and preload. Digoxin may be used to improve left ventricular function following consultation with a cardiologist. Digoxin has potential risk in patients with increased myocardial sensitivity secondary to the inflammatory changes of myocarditis. Inotropic agents, such as dopamine and dobutamine may be necessary if hemodynamic instability is present. Afterload-reducing agents help reduce the workload for the poorly functioning myocardium. If hypotension is not present, intravenous nitroprusside or milrinone may be used. Oral agents such as ACE inhibitors may be used if the patient is stable.
Intravenous gamma globulin in the setting of acute myocarditis may improve ventricular function and survival.44 The use of immunosuppressive agents (e.g., steroids or cyclosporine) in acute myocarditis is controversial, and therefore should not be initiated in the ED. All patients with myocarditis should be admitted to a PICU for further management and observation.
The diagnosis of myocarditis may not be made in the ED; endomyocardial biopsy is the gold standard for diagnosis of myocarditis. Patients may present with a seemingly benign viral illness with symptoms of weakness, fever, and emesis; it is important for the clinician to maintain a high degree of suspicion for this disease.
Pericarditis. The pericardium consists of two layers: the visceral layer and the parietal layer. In a healthy person, a small amount (10-15 mL in a child) of serous fluid normally exists. When inflammation of the pericardium occurs, fluid accumulates between the two layers of the pericardium and forms a pericardial effusion. Normally, the pericardium does not affect the filling of the heart. However, when a pericardial effusion is present, the filling capacities of the heart chambers are limited, resulting in increased end-diastolic filling pressures and decreased cardiac output. Acute development of an effusion is more likely to result in such complications because the pericardium does not have time to stretch and accommodate the increased volume. Significant effusions may lead to cardiac tamponade.
The frequency of pericarditis is unknown, and there does not appear to be any sex or age predilection. Symptoms vary depending on the etiology and how rapidly the pericardial effusion develops. Common presenting symptoms include respiratory difficulty, fever, and substernal chest pain that may radiate to the left shoulder. The chest pain may be accentuated by lying flat or respiratory motion and improved with sitting forward. Patients with a viral etiology may have a recent history of illness, including upper respiratory tract symptoms and fever. There are both infectious and noninfectious causes of pericarditis. Among the infectious causes, viral etiologies (e.g., coxsackievirus A and B, ECHO viruses, and influenza) are more common.
Physical findings vary depending on the severity of disease. With no significant effusion, the only sign on physical examination may be a friction rub. With more serious disease, there will be tachypnea and tachycardia. Signs of cardiac tamponade include distended neck veins, clear lungs, weak peripheral pulses, tachycardia, distant heart tones with auscultation, and a pulsus paradoxus.
A CBC count may reveal an elevated white blood cell count in the presence of bacterial pericarditis, but may be normal with other etiologies. Measurements of electrolyte, BUN, and creatinine levels are indicated. Appropriate cultures (e.g., blood, urine, stool) and viral serology should be obtained, and an erythrocyte sedimentation rate (ESR) may be helpful.
The ECG typically shows diffuse ST-segment elevation in both the limb and precordial leads with associated upright T-waves. ST depression in aVR and PR-segment depression also may be present. (See Figure 4.) With chronic pericarditis, the ST-segment elevation resolves and widespread T-wave inversion develops. A large effusion may be associated with generalized low voltage and electrical alternans on ECG, although the latter is an uncommon finding.
The initial chest X-ray study usually demonstrates clear lung fields and a cardiac silhouette that may be mistaken for cardiomegaly. A large effusion may give the appearance of a water-bottle shaped heart. An echocardiogram is the diagnostic study of choice and demonstrates the effusion, quantifies the amount and thickness of the pericardium, and allows evaluation of the cardiac hemodynamic function.
Advanced techniques of non-surgical percutaneous pericardial biopsy, pericardioscopy, cytologic analysis of pericardial fluid, and molecular techniques (e.g., polymerase chain reaction and in situ hybridization) currently are available.45-47 Such techniques significantly improve diagnosis of specific etiologies, but are not available immediately in the ED.
Following a cardiology consultation, stable patients with idiopathic or viral pericarditis may be treated as outpatients with nonsteroidal anti-inflammatory agents. Salicylates can be used, as can ibuprofen.48 The patient should be placed on bed rest for 1-3 weeks with close follow-up. In stable patients with no signs of decompensation, pericardiocentesis may be deferred safely, particularly if removal of fluid is for diagnostic purposes only. In an unstable patient with evidence of tamponade, pericardiocentesis should be performed immediately. In addition, an initial fluid bolus should be given if hypotension is present. Antibiotics should be initiated until a bacterial source can be excluded. Recommended antibiotics include nafcillin and cefotaxime.48 Corticosteroids have been found effective only with certain etiologies of pericarditis (i.e., autoreactive).49 Steroids should not be used until a bacterial etiology can be excluded, and are not recommended in acute viral pericarditis. A pediatric cardiology consult is required, and all unstable patients should be admitted to an ICU setting.
Congestive Heart Failure (CHF)
CHF can be defined as an inability of cardiac output to meet the metabolic needs of the body. The causes of CHF in children tend to differ from those in adults. In developed countries, the main etiologies of CHF in children include congenital heart defects, cardiomyopathy, and myocardial dysfunction following heart defect repair.50 Of these three etiologies, congenital heart defects are the most common. The incidence of congenital heart defects is approximately eight of every 1,000 live births.33 Among children with congenital heart defects, the prevalence of CHF can be as high as 20%.51 Cardiomyopathy has multiple etiologies including genetic neuromuscular and metabolic disorders, collagen vascular diseases, hypothyroidism, hyperthyroidism, viral infections, drugs (e.g., anthracycline and cyclophosphamide), malnutrition, and hematologic disorders (e.g., sickle cell anemia). Studies have shown an annual incidence of cardiomyopathy ranging from 0.34-0.6 cases per 100,000 persons.52,53 The prevalence of CHF following heart defect repair is unclear and appears to depend on the specific defect and repair performed.
Common symptoms of CHF in infancy include poor feeding, a prolonged feeding time, an increased respiratory rate and effort, and excessive sweating.54,55 Physical exam findings for an infant with CHF classically include pallor, tachycardia, tachypnea, diaphoresis, and hepatomegaly. An S3 gallop rhythm may be present. A sternal heave and laterally displaced point of maximal impulse indicates cardiomegaly. Rales are not always heard on auscultation of the lungs, and the absence of rales does not exclude the possibility of CHF. Peripheral edema is a very uncommon finding in infants. Failure to thrive in an infant is a hallmark of heart failure.55
Older children with heart failure often have poor exercise tolerance and fatigue in addition to poor appetite, growth failure, and increased respiratory rate and effort.50 Atypical symptoms (e.g., abdominal pain, nausea, weight loss, and anorexia) also may occur in older children.55 Signs of CHF in older children include tachycardia, tachypnea, rales on auscultation of the lungs, and hepatomegaly. Jugular venous distention and peripheral edema also may be present.
Laboratory tests in the ED should include a CBC count, and measurements of electrolytes, BUN, creatinine, and blood glucose levels. Hyponatremia may be present with fluid overload. An ABG measurement may help evaluate oxygenation and identify respiratory acidosis. Cardiac enzymes are indicated if ischemia or myocarditis is suspected.
A chest x-ray should be performed in all children evaluated for CHF. The chest x-ray helps identify pulmonary vascular congestion and cardiomegaly. A cardiothoracic ratio on the chest x-ray greater than 0.55 indicates cardiomegaly in infants, and a ratio greater than 0.5 is indicative in children older than 1 year.56,57
Other essential tests include an ECG and echocardiogram. The ECG may help identify causes of CHF, such as myocardial ischemia or dysrhythmias, or it may be nonspecific. It also can help identify heart chamber enlargement and electrolyte abnormalities. A low voltage in the QRS-complex may suggest myocarditis. Echocardiography is valuable in identifying structural lesions, valve abnormalities, potential effusions, ventricular wall motion abnormalities, and assessing the ejection fraction.
As in other situations involving unstable patients, or potentially unstable patients, the first priorities of management are assessment and stabilization of the patient’s airway, breathing, and circulation. The patient should be provided supplemental oxygen, and ventilation assisted as necessary. Cardiac monitoring, pulse oximetry, and frequent blood pressure measurements should be initiated immediately and obtained regularly to assess for changes in hemodynamic status. Agents such as dopamine or dobutamine may be required for cardiovascular support. At low doses (2-5 mcg/kg/min), dopamine improves renal perfusion. Moderate doses of dopamine (5-10 mcg/kg/min) increase myocardial contractility via beta1-adrenergic receptor activation, and evoke vasoconstriction through alpha-adrenergic receptor effects. Doses greater than 10 mcg/kg/min have predominantly vasoconstrictive effects. The improvements in hemodynamic status with dopamine come at the expense of tachycardia, increased myocardial oxygen demand, and the risk of tachydysrhythmias. Dobutamine (2-15 mcg/kg/min) primarily functions as an inotrope and vasodilator. Secondary to the vasodilatory effects, dobutamine may not increase blood pressure significantly, and actually decreases peripheral resistance. Therefore, if significant hypotension is present, dobutamine should not be the primary inotrope initiated. Furosemide (1mg/kg IV) reduces preload and decreases fluid overload in patients with pulmonary edema. In some situations, vasodilators may help by decreasing afterload to improve cardiac output. Nitroprusside (0.5-10 mcg/kg/min IV) has both venous and arterial effects and provides afterload reduction and venodilation. Intravenous nitroglycerin also may be used, but is primarily a venodilator.
In stable patients, following consultation with a cardiologist, digoxin is the indicated drug for inotropic support.8,48 Digoxin acts to decrease the heart rate, increase myocardial contractility, and decreases sympathetic outflow.58 The initial loading dose, or total digitalizing dose (TDD), is given during a 24-hour period, with half of the total dose given initially, then a quarter of the total dose given 8-12 hours after the first dose, and the remaining quarter dose given 8-12 hours after the second dose.8,48 The oral TDD in full-term newborns is 30 mcg/kg, and in children older than 1 year the TDD is 30-50 mcg/kg. It is important to note that the intravenous TDD is only 75% of the oral TDD.
Other agents like angiotensin-converting enzyme (ACE) inhibitors have been shown to improve survival in adults with CHF and may have some benefit in children with CHF.59 However, these agents generally are used for chronic maintenance and not in the ED setting.
Conclusions
Cardiac disorders are uncommon in children, and present infrequently to the ED. However, as illustrated in this discussion, significant morbidity and mortality are associated with these conditions if not recognized and treated promptly. Cardiac diseases must be considered in the differential of infants who present with acute illnesses in the first month of life and older children with suggestive signs and symptoms, and particularly children who appear ill and fail to respond to common management. Early recognition and aggressive management may significantly improve the child’s chance for an optimal outcome.
References
1. Hazinski MF, Zaritsky AL, Nadkarini VM, et al, eds. PALS Provider Manual. Dallas: American Heart Association, 2002.
2. Case C. Diagnosis and treatment of pediatric arrhythmias. Pediatr Clin N Amer, 1999;46:347-354.
3. O’Laughlin MP. Congestive heart failure in children. Pediatr Clin North Am. 1999;46:263-273.
4. Danford DA, Kugler JD, Deal B, et al. The learning curve for radiofrequency ablation of tachyarrhythmias in pediatric patients. Am J Cardiol 1995;75:587-590.
5. Luedtke SA, Kuhn RJ, McCaffrey FM. Pharmacologic management of supraventricular tachycardias in children. Part 1: Wolff-Parkinson-White and atrioventricular nodal reentry. Ann Pharmacother 1997;31:1227-1243.
6. Perry JC, Garson A Jr. Supraventricular tachycardia due to Wolff- Parkinson-White syndrome in children: Early disappearance and late recurrence. J Am Coll Cardiol 1990;16:1215-1220.
7. Perry JC. Supraventricular Tachycardia. In: Garson A, Bricker JT, Fisher DJ, et al. The Science and Practice of Pediatric Cardiology. Baltimore: Williams & Wilkins; 1998.
8. Gewitz MH, Vetter VL. Cardiac Emergencies. In: Fleisher GR, Ludwig, S eds. Textbook of Pediatric Emergency Medicine, 4th ed. Philadelphia: Lippincott Williams & Williams;2000.
9. Fitzmaurice L, Gerardi MJ. Cardiovascular System. In: Gausche-Hill, M, Fuchs, S, Yamamoto L, eds. The Pediatric Emergency Medicine Resource, 4th ed. Sudbury: Jones and Bartlett Publishers;2004.
10. Chameides L. Cardiovascular Disorders. In: Barkin RM, ed. Pediatric Emergency Medicine, Concepts and Clinical Practice. 2nd ed., St. Louis: Mosby, Inc; 1997.
11. Ackerman MJ. The long QT syndrome: Ion channel diseases of the heart. Mayo Clin Proc 1998;73:250-269.
12. Garson AJ, Dick M, Fournier A, et al. The long QT syndrome in children: An international study of 287 patients. Circulation 1993;87: 1866-1872.
13. Moss AJ. Prolonged QT interval syndromes. JAMA 1986;256: 2985-2987.
14. Haslam RHA. The nervous system. In: Nelson WE, ed. Textbook of Pediatrics. Philadelphia: WB Saunders;2000:1830.
15. Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998;338:1709-1714.
16. Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long QT syndrome. N Engl J Med 2000;343:262-267.
17. Bazett HC. An analysis of the time-relations of electrocardiograms. Heart 1920;7:353-370.
18. Moss AJ. Measurement of the QT interval and the risk associated with QTc interval prolongation: A review. Am J Cardiol 1993;72: 23-25.
19. Friedman J, Mull C, Sharieff G. Prolonged QT Syndrome in children: An uncommon but potentially fatal entity. J Emerg Med, in press.
20. Miller MD, Porter CB, Ackerman MJ. Diagnostic accuracy of screening electrocardiograms in long QT syndrome I. Pediatrics 2001;108: 8-12.
21. Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J 1985;109:399-411.
22. O’Connor BK. Arrhythmias in long QT and WPW syndromes. In: Gillette PC, Garson A, eds. Clinical Pediatric Arrhythmias. Philadelphia: WB Saunders, 2nd edition;1999:306-314.
23. Salen P, Nadkarni V. Congenital long QT syndrome: A case report illustrating diagnostic pitfalls. J Emerg Med 1999;17:859-864.
24. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol 1992;20:1391-1396.
25. Plunkett A, Hulse JA, Mishra B, et al. Variable presentation of Brugada syndrome: Lessons from three generations with syncope. BMJ 2003;326:1078-1079.
26. Mattu A, Rogers RL, Kim H, et al. The Brugada syndrome. Am J Emerg Med 2003;21:146-151.
27. Priori SG, Napolitano C, Giordano U, et al. Brugada syndrome and sudden cardiac death in children. Lancet 2000;355:808-809.
28. Brugada P, Brugada R, Brugada J. The Brugada syndrome. Curr Cardiol Rep 2000;2:507-514.
29. Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002;105:1342-1347.
30. Wilde AAM, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation 2002;106: 2514-2519.
31. Brugada P, Brugada R, Mont L, et al. Natural history of Brugada syndrome: The prognostic value of programmed electrical stimulation of the heart. J Cardiovasc Electrophysiol 2003;14:455-457.
32. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003;108: 3092-3096.
33. McCollough M, Sharieff G. Common complaints in the first 30 days of life. Emerg Med Clin North Am 2002;20:27-48.
34. Woolridge D, Love J. Congenital heart disease in the pediatric emergency department. Pathophysiology and clinical characteristics. Pediatr Emerg Med Rep 2002;7;69-80.
35. Zahka K. Approach to the neonate with cardiovascular disease. In: Avroy F, Martin R, eds. Neonatal-perinatal medicine. Diseases of the fetus and infant. St Louis: Mosby;1997:1119-1137.
36. Flynn PA, Engle MA, Ehlers KH et al. Cardiac issues in the pediatric emergency. Pediatr Clin North Am 1992;39:955-983.
37. DiMaio A, Singh J et al. The infant with cyanosis in the emergency department. Pediatr Clin North Am, 1992;39;987-1006.
38. Savitsky E, Alejos J, Votey S. Emergency department presentations of pediatric congenital heart disease. J Emerg Med, 2003;24:239-245.
39. Kay JD, Colan SP, Graham TP. Congestive heart failure in pediatric patients. Am Heart J 2001;142:923-928.
40. Cotran RS, Kumar V, Robbins SL. Robbins Pathologic Basis of Disease. 4th ed. Philadelphia; WB Saunders:1989.
41. Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: Clinical spectrum and treatment. Circulation 1995;92: 1680-1692.
42. Jouriles N. Pericardial and myocardial disease. In: Marx J, Hockberger R, Walls R, eds. Rosen’s Emergency Medicine: Concepts and Clinical Practice. St. Louis: Mosby;2002:1143-1144.
43. Kopecky SL, Gersh BJ. Dilated cardiomyopathy and myocarditis: Natural history, etiology, clinical manifestations, and management. Curr Probl Cardiol 1987;12:569-647.
44. Drucker NA, Colan SD, Lewis AB, et al. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation 1994;89: 252-257.
45. Maisch B. Pericardial diseases, with a focus on etiology, pathogenesis, pathophysiology, new diagnostic imaging methods, and treatment. Curr Opin Cardiol 1994;9:379-388.
46. Uthaman B, Endrys J, Abushaban L, et al. Percutaneous pericardial biopsy: Technique, efficacy, safety, and value in the management of pericardial effusion in children and adolescents. Pediatr Cardiol 1997;18:414-418.
47. Maisch B, Ristic AD, Seferovic PM. New directions in diagnosis and treatment of pericardial disease. A project of the Taskforce on Pericardial Disease of the World Heart Federation, Herz 2000;25: 769-780.
48. Li MM, Klassen TP, Watters LK. Cardiovascular Disorders. In: Pediatric Emergency Medicine, Concepts and Clinical Practice. 2nd ed. St. Louis: Mosby, Inc.; 1997.
49. Maisch B, Ristic AD. The classification of pericardial disease in the age of modern medicine. Curr Cardiol Rep 2002;4:13-21.
50. Kay JD, Colan SD, Graham TP. Congestive heart failure in pediatric patients. Am Heart J 2001;142:923-928.
51. Buchhorn R, Hulpke-Wette M, Hilgers R, et al. Propranolol treatment of congestive heart failure in infants with congenital heart disease: The CHF-PRO-INFANT Trial. Int J Cardiol 2001; 79:167-173.
52. Arola A, Jokinen E, Ruuskanen O, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland, Am J Epidemiol 1997;46:385-393.
53. Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy: The Prospective Pediatric Cardiomyopathy Registry. J Am Coll Cardiol 2001;37:465-466.
54. Ross RD, Bollinger RO, Pinsky WW. Grading the severity of congestive heart failure in infants. Pediatr Cardiol 1992;13:72-75.
55. Clark BJ 3rd. Treatment of heart failure in infants and children. Heart Dis 2000;2:354-361.
56. Artman M, Graham TP. Congestive heart failure in infancy: Recognition and management. Am Heart J 1982;203:1040-1055.
57. Artman M, Parrish MD, Graham TP. Congestive heart failure in childhood and adolescence: Recognition and management. Am Heart J 1983;204:471-480.
58. Venugopalan P, Agarwal AK, Worthing EA. Chronic cardiac failure in children due to dilated cadiomyopathy: Diagnostic approach, pathophysiology and management. Eur J Pedatri 2000;159:803-810.
59. Shaddy RE. Optimizing treatment for chronic congestive heart failure in children. Crit Care Med 2001;29:S237-S240.
Although pediatric cardiac diseases infrequently are seen in the emergency department (ED), early diagnosis and aggressive management is critical. Most importantly, the clinician must include these diseases in their differential and have a thorough understanding of typical and atypical presentations for congenital heart disease, dysrhythmias, myocarditis and pericarditis. Any child who has a clinical presentation suggestive of cardiac disease, must receive appropriate diagnostic testing and timely referral to optimize the childs outcome. The authors provide a thorough, focused review of the most commonly encountered cardiac diseases in the ED and key aspects to stabilization.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.