The Role of RET in Pheochromocytoma
The Role of RET in Pheochromocytoma
By Christian A. Koch, MD, Karel Pacak, MD, PhD,
Steve C. Huang, PhD, Alexander O. Vortmeyer, MD, and Zhengping Zhuang, MD, PhD
Proto-oncogenes and oncogenes such as met and RET play important roles in the development of tumors. Often, however, the exact mechanisms of tumorigenesis are unknown. There are many in vitro studies and animal models on the study of tumor formation in oncogene-driven disorders, but only a few, recent in vivo studies on hereditary tumor syndromes. Zhuang and associates reported on hereditary papillary renal carcinomas associated with germline mutations of MET and showed that a subset of these tumors develop by duplication of mutant MET in trisomy 7.1 Based on these findings, in multiple endocrine neoplasia type 2 (MEN 2)-related pheochromocytomas Huang and Koch tested whether RET, another proto-oncogene with structural and functional homology to MET, would lead to tumorigenesis through a similar mechanism.2 This work will elucidate tumor formation in MEN 2-related tumors and may lead to a better understanding of tumorigenesis of other oncogene-related hereditary tumor syndromes.
Background
Pheochromocytoma is a neuroendocrine tumor that often leads to paroxysmal hypertension, headaches, stroke, cardiac arrhythmias, and so-called spells.3 Most of these tumors occur in the sporadic form, but about 10% are hereditary. Among hereditary syndromes, pheochromocytoma most commonly occurs in MEN 2, followed by von Hippel-Lindau disease and neurofibromatosis type 1. The gene responsible for MEN 2 is the RET proto-oncogene.
RET has been mapped to chromosome 10q11.2 and subsequently has been identified.4-6 Its name stems from transfection studies and stands for "rearranged during transfection." RET consists of 21 exons, with six exons called "hot spots." These hot spots are areas where germline mutations of RET frequently are detected in patients with MEN 2. Germline mutations of RET are responsible for the familial tumor syndrome, MEN 2, which is subdivided into three groups: familial medullary thyroid carcinoma; MEN 2A, including medullary thyroid carcinoma, pheochromocytoma, and parathyroid hyperplasia/adenoma; and MEN 2B, consisting of medullary thyroid carcinoma, pheochromocytoma, and mucosal and other neuromas, as well as certain body features such as a marfanoid body habitus. RET encodes a receptor tyrosine kinase, the ligands of which are glial cell line-derived neurotrophic factor (GDNF) and neurturin.4 GDNF is a member of the transforming growth factor (TGF)-b family. RET activation by GDNF appears to occur via a membrane-bound protein, GFRa, which seems to function as the ligand-binding domain of the ligand-receptor complex.
Tumorigenesis in MEN 2-Related Pheochromocytoma
Although it has been known that patients with RET germline mutations develop hyperplasia of the parafollicular C-cells in the thyroid gland and of the adrenal medulla, the mechanisms of tumor formation in patients with MEN 2-related medullary thyroid carcinoma and pheochromocytoma are widely unknown. Whereas tumor suppressor genes such as VHL are believed to initiate tumorigenesis according to Knudson’s two-hit model,7 oncogenes such as RET may lead to tumor formation by other mechanisms. In a recent study, two possible mechanisms of tumorigenesis in MEN 2-related pheochromocytoma have been suggested.2
Hereditary tumor syndromes are ideal for studying the mechanisms of tumor formation. Huang and Koch investigated nine pheochromocytomas from five un-
related patients with MEN 2.2 By performing micro-
dissection of frozen tumor tissue, fluorescent in situ hybridization, quantitative PCR, linkage analyses, phosphorimage densitometry, and mutation analyses by single-strand conformation polymorphism and restriction enzyme digestion, they found that these pheochromocytomas showed duplication of the mutant RET allele or loss of the wild-type allele (see Figure). These results indicate a dominant effect of mutant RET and suggest this effect as a mechanism of tumor formation in MEN 2-related pheochromocytoma. This dominant effect of RET could represent the so-called "second hit" in tumor formation analogous to the two-hit model in tumor suppressor gene-associated tumors.7 In ongoing studies, researchers found that patients with MEN 2 might have more than one RET mutation.8-10 In this scope, however, Koch and Huang also discovered that somatic RET mutations in pheochromocytomas from patients with MEN 2 likely represent a phenomenon of tumor progression (unpublished data). The biological/functional significance of more than one RET mutation in patients with MEN 2 must be elucidated.
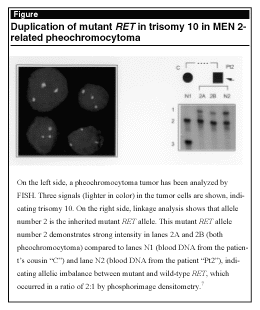 |
Analysis of RET mutations in sporadic pheochromocytomas has revealed somatic mutations in up to 15% of tumors, with the codon 918 somatic RET mutation (MEN 2B mutation) as the one most commonly found (in 3-10% of cases).11-15 On the other hand, exon 10 and 11 somatic RET mutations (as seen in MEN 2A) occur in less than 1% of tumors. Based on this low number of somatic RET mutations in sporadic pheochromocytomas and our findings in MEN 2-related pheochromocytoma, we propose that other mechanisms of tumor formation, in addition to a somatic RET mutation, must play a role in the development of sporadic pheochromocytomas.
Conclusion
Pheochromocytomas are neuroendocrine tumors that occur most often in the adrenal glands. Duplication of mutant RET in trisomy 10 or loss of wild-type RET in MEN 2-related pheochromocytoma can lead to a dominant effect of mutant RET. This effect likely represents the "second hit" of tumor formation, the first hit being hyperplasia of the adrenal medullary cells caused by the effect of one mutant RET allele. Further studies are needed to elucidate mechanisms of tumor formation in pheochromocytoma. (Drs. Koch and Pacak are Clinical Investigators, Pediatric Reproductive and Endocrinology Branch, National Institute of Child Health and Hu-man Development, NIH; Dr. Vortmeyer is Staff Attending, Neuropathology, Dr. Huang is Research Fellow, and Dr. Zhuang is Head, Molecular Pathogenesis Unit, Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, NIH, Bethesda, MD.)
References
1. Zhuang Z, Park WS, Pack S, et al. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet 1998;20:66-69.
2. Huang SC, Koch CA, Vortmeyer AO, et al. Duplication of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine neoplasia type 2-associated pheochromocytoma. Cancer Res 2000;60:
6223-6226.
3. Pacak K, Chrousos GP, Koch CA, et al. Pheochromocytoma: Progress in diagnosis, therapy, and genetics. In: Chrousos GP, Margioris AN, eds. Adrenal Disorders. New York: Humana Press; 2001:379-413.
4. Ponder BA. The phenotypes associated with RET mutations in the multiple endocrine neoplasia type 2 syndrome. Cancer Res 1999;59:1736s-1742s.
5. Eng C. RET proto-oncogene in the development of human cancer. J Clin Oncol 1999;17:380-393.
6. Gagel RF. Multiple endocrine neoplasia type 2. In: DeGroot, LJ, Jameson L, eds. Endocrinology. Orlando, FL: WB Sauders Co.; 2001:2518-2532.
7. Knudson AG. Genetics of human cancer. Annu Rev Genet 1986;20:231-251.
8. Koch CA, Huang SC, Vortmeyer AO, et al. A patient with MEN 2 and multiple mutations of RET in the germline. Exp Clin Endocrinol Diabetes 2000;108:493.
9. Bartsch DK, Hasse C, Schug C, et al. A RET double mutation in the germline of a kindred with FMTC. Exp Clin Endocrinol Diabetes 2000;108:128-132.
10. Tessitore A, Sinisi AA, Pasquali D, et al. A novel case of multiple endocrine neoplasia type 2A associated with two de novo mutations of the RET protooncogene. J Clin Endocrinol Metab 1999;84:3522-3527.
11. Lindor NM, Honchel R, Khosla S, et al. Mutations in the RET protooncogene in sporadic pheochromocytomas. J Clin Endocrinol Metab 1995;80:627-629.
12. Beldjord C, Desclaux-Arramond F, Raffin-Sanson M,
et al. The RET protooncogene in sporadic pheochromocytomas: Frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab 1995;80:
2063-2068.
13. Bar M, Friedman E, Jakobovitz O, et al. Sporadic pheochromocytomas are rarely associated with germline mutations in the von Hippel-Lindau and RET genes. Clin Endocrinol (Oxford) 1997;47:707-712.
14. Eng C, Crossey PA, Mulligan LM, et al. Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. J Med Genet 1995;32:
934-937.
15. Brauch H, Hoeppner W, Jahnig H, et al. Sporadic pheochromocytomas are rarely associated with germline mutations in the VHL tumor suppressor gene or the RET protooncogene. J Clin Endocrinol Metab 1997;82:4101-4104.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.