Congenital Bleeding Disorders: Diagnostic Features and Syndrome-Specific Management in the Emergency Department
Congenital Bleeding Disorders: Diagnostic Features and Syndrome-Specific Management in the Emergency Department
Authors: David A. Wald, DO, Assistant Residency Director, Co-Director Emergency Medicine Clerkship, Division of Emergency Medicine, Temple University Hospital, Philadelphia, PA; Paul Alleyne, MD, Division of Emergency Medicine, Temple University Hospital, Philadelphia, PA.
Peer Reviewer: Sandra M. Schneider, MD, FACEP, Professor and Chair, Department of Emergency Medicine, University of Rochester, Rochester, NY.
Von Willebrand’s disease (vWD) is the most commonly encountered inherited bleeding disorder.1-6 It is estimated that vWD affects at least 1% of the general population, and has a prevalence of 1 in 1000 live births.1,2,4,6 Although several rare autosomal recessive variants have been identified, vWD usually is inherited as an autosomal dominant trait affecting men and women equally without an ethnic predominance.7,8
The hemophilias constitute a group of sex-linked recessive disorders affecting males almost exclusively. Although uncommon, hemophilia has been documented in a minority of female carriers, and may occur as a result of a spontaneous gene mutation that might be responsible for up to 30% of all new cases.8,9 Together, hemophilias A, B, and C (factor VIII, IX, and XI deficiencies) account for 99% of all inherited bleeding disorders that result from coagulation factor deficiencies.10 It is estimated that about 20,000 individuals in the United States have hemophilia. The prevalence of hemophilia A is approximately 1 in every 5000 live male births, and is about four times more common than hemophilia B.1,8,11,12 By the mid 1980s, the life expectancy of patients with severe hemophilia approached 62 years.8 In the last decade, acquired immune deficiency syndrome (AIDS) has emerged as the leading cause of death in patients with hemophilia.8 Prior to the initiation of viral inactivation of all factor concentrates in 1985, 55% of all persons with hemophilia were infected with the human immunodeficiency virus (HIV).12 More than 4000 patients have since progressed to AIDS, and as of June 1994, more than 2500 have died.13 The United States Hemophilia/HIV Seroconversion Project has found no cases of HIV seroconversion that are attributable to factor concentrate administration since 1987, and no cases of hepatitis B or C seroconversion since 1992.13 The last decade also has brought technological advances, including monoclonal antibody purification and recombinant technology that have further improved the efficacy of hemophilia treatment. This has not occurred without a price; severely affected adults may require 50,000-80,000 IU of factor concentrate annually, with a cost of up to $100,000.8
Emergency medicine physicians play a primary role in the acute management of the hemorrhagic manifestations of patients with congenital bleeding disorders. Early intervention and aggressive factor replacement often is required to limit the morbidity and mortality associated with these conditions. This review will highlight the most common inherited bleeding disorders an emergency medicine physician will likely encounter, and provide a concise and logical approach to their treatment.—The Editor
Normal Mechanisms of Hemostasis
In response to a vascular injury, a detailed sequence of events is triggered by the body that lead to hemorrhage control. Hemostasis occurs as a result of complex interactions between the vascular subendothelium, platelets, and various plasma coagulation factors. To better understand the role of these three components in the process, a brief review is in order. A modified approach follows that can be used to better conceptualize the various mechanisms responsible for coagulation.14
Primary hemostasis refers to the interactions that occur between the exposed subendothelium of an injured blood vessel and circulating platelets. Immediately following the loss of vascular integrity, transient arteriolar vasoconstriction occurs. This largely occurs as a result of reflex mechanisms and is enhanced by the release of tissue thromboplastin from the injured vascular endothelium.14,15 Tissue thromboplastin also is responsible for initiating the extrinsic pathway of the coagulation cascade system. The injured endothelium provides a surface (collagen) that promotes platelet adhesion and initiates a series of steps responsible for the formation of a primary hemostatic plug. von Willebrand factor (vWF) is a large multimeric glycoprotein that plays a key role in platelet adhesion by binding with exposed subendothelial collagen and platelet receptor glycoprotein Ib.1,2,7,8,14,16 Further platelet activation and aggregation occur as a result of fibrinogen and vWF binding to the glycoprotein IIb/IIIA platelet receptor.3,6,17 Platelet activation also promotes the exposure of a platelet membrane phospholipid layer that can provide a contact surface and help initiate the intrinsic pathway of the coagulation cascade system.14,15
Secondary hemostasis refers to a complicated and often interrelated set of pathways known as the coagulation cascade system. Traditional teaching divides the coagulation cascade into the extrinsic and intrinsic pathways. This division allows one to better conceptualize these two systems. Although often discussed as distinct pathways, in vivo, it is apparent that there are interconnections between these two pathways.14,15 A simplified overview of the coagulation cascade system can be seen in Figure 1.
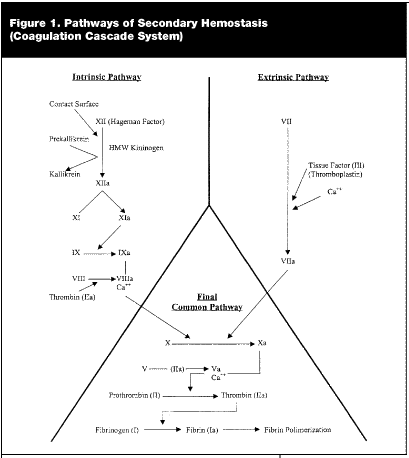 |
vWF also plays an important role in secondary hemostasis. By acting as a carrier protein (factor VIII:vWF complex), vWF protects factor VIII from proteolytic degredation.4,6,17 When plasma vWF levels are normal, the circulating half-life of factor VIII is approximately 12 hours; when a qualitative or quantitative defect of vWF occurs, the half-life of factor VIII can decrease to 2.4 hours.7
Clinical and Laboratory Characteristics of Inherited Bleeding Disorders
The assessment of a patient with a suspected bleeding disorder should include a detailed family history and a description of prior bleeding episodes. A family history of a known or suspected coagulopathy may be helpful, but a negative history does not exclude the possibility of an inherited bleeding disorder. Various underlying medical conditions, such as uremia, liver disease, certain myeloproliferative diseases, or the use of antiplatelet agents, may result in an acquired bleeding disorder.
Although overlapping features occur, a clinical distinction often can be made between disorders of coagulation (factor deficiencies) and disorders of platelets or blood vessels. Disorders of coagulation most frequently are associated with the development of large superficial ecchymosis, muscular hematomas, and prolonged traumatic or post-surgical hemorrhage. Disorders of platelets or blood vessels more commonly are associated with mucocutaneous bleeding.
The prothrombin time (PT) is the most commonly ordered screening coagulation function test.14 In vitro, this test activates the extrinsic coagulation cascade and is an indicator of defects of factors VII, X, and V; prothrombin; and fibrinogen (extrinsic and final common pathways). Three of these factors are vitamin K-dependent (prothrombin and factors VII and X). The PT is more sensitive to factor VII deficiencies because of the short half-life of this coagulation protein (4-6 hours).9 In addition to various factor deficiencies, prolongation of the PT commonly is associated with cirrhotic liver disease, and commonly is used to monitor the effectiveness of coumadin therapy.
The activated partial thromboplastin time (aPTT) evaluates the integrity of the intrinsic and final common pathway (high molecular weight kininogen [HMWK]; prekallikrein; factors XII, XI, IX, VIII, X, and V; prothrombin; and fibrinogen). The aPTT will be prolonged when deficiencies of these various factors occur (< 30% of normal), and commonly will be used to evaluate the effectiveness of heparin therapy.
The bleeding time is a sensitive, although nonspecific, indicator of the adequacy of primary hemostasis. The template bleeding time is the standard used, although some laboratories will use variations of this technique.18 This is performed by making a standardized incision on the volar aspect of the forearm and determining the time required for the bleeding to stop. Mild thrombocytopenia usually does not affect the bleeding time, but when the platelet count is less than 100,000/mm3, there is correlation between the degree of prolongation and the severity of the thrombocytopenia.18 Other defects in platelet function, such as Glanzmann’s thrombasthenia, uremia, or myeloproliferative disorders, can prolong the bleeding time.16,18 Coagulation factor deficiencies commonly do not prolong the bleeding time.
vWD often is associated with a prolonged bleeding time, a normal platelet count, and a normal PT and aPTT. In mild cases, the bleeding time measurement may be within normal limits; in severe cases, the aPTT may be prolonged if the factor VIII level is less than 30%. A more in-depth hematologic workup is required to confirm the diagnosis of vWD.
Hemophilia A (factor VIII deficiency) does not affect the platelet count or the PT. The bleeding time will be elevated in up to 20% of cases, and occurs more often in cases of severe disease.18 The aPTT is the screening test of choice for these patients as it will be prolonged in virtually all cases of moderate and severe disease. The aPTT may not be sensitive enough to identify all mild cases and some carrier states. Routine laboratory abnormalities commonly associated with hemophilia B are similar to those seen with hemophilia A. Factor XI is considered one of four contact activation factors. Factor XI is responsible for activating factor IX in the intrinsic coagulation pathway; deficiency of this factor also will prolong the aPTT. Deficiencies of the other three contact factors (factor XII, prekallikrein, and HMWK) will prolong the aPTT, but are not associated with clinical bleeding.8 A more in-depth evaluation, including coagulation factor assay, often is required to diagnose specific factor deficiencies.
von Willebrand’s Disease
First described by Erik Adolf von Willebrand in 1926, vWD causes a defect in primary hemostasis. In 1994, a new classification system was devised for vWD.19 This new system reclassifies the defect by the mechanism of bleeding and helps to incorporate the more than 20 subtypes that have been identified previously into three larger, more inclusive groupings. Quantitative defects of vWF are divided into mild-to-moderate deficiency (type 1) and severe deficiency (type 3); qualitative deficiencies (type 2) are further divided into four subtypes. This reclassification also is helpful because treatment options are, in part, determined by the type or subtype of vWD. Bleeding complications associated with vWD are variable and will depend on the subtype and the severity of the disease. Even within affected families, the frequency and severity of bleeding complications will vary.4
Type 1 vWD is the most common form of the disease and occurs in about 75-80% of affected patients.2-4,17 Individuals with this type of disease have a mild-to-moderate decrease in plasma levels of vWF. Although variable, these patients often have a mild-to-moderate bleeding diathesis. The vWF activity is reduced to 20-50% of normal, and patients will have a proportional decrease in circulating factor VIII levels.3,20
Type 2 vWD occurs in about 15-20% of affected patients.2,17 This type encompasses a variety of qualitative defects of vWF. Type 2A, which is the most common type 2 variant, occurs in about 10-15% of cases.3 Patients with this variant lack the high molecular weight multimers (HMWMs) of vWF that are required to mediate platelet adhesion.4 Type 2B, including platelet—type pseudo—vWD is rare and occurs in less than 5% of cases. In this subtype, excessive binding of vWF to platelet glycoprotein Ib occurs, resulting in platelet activation and removal of vWF from circulation. This interaction will lead to decreased plasma concentrations of vWF.4
Type 2M is an extremely rare variant in which platelet dysfunction occurs as a result of a qualitative defect in vWF, not affecting the HMWMs. Type 2N also is very rare, and results from a defect in the vWF-factor VIII binding region. As a result of this defect, the unbound factor VIII undergoes more rapid proteolytic degredation.
Type 3 vWD is a severe bleeding disorder that occurs in less than 1% of affected patients. Individuals with this subtype have essentially no detectable plasma levels of vWF, and will have proportional decreases in plasma levels of factor VIII.
Many patients with type 1 vWD (mild) will be asymptomatic unless challenged by surgery or trauma. The clinical manifestations associated with vWD occur as a result of a defect in primary hemostasis (platelet adhesion and activation) and generally lead to mucocutaneous bleeding. Common presenting complaints are easy bruising and mucous membrane bleeding, such as epistaxis, gingival, gastrointestinal (GI), and vaginal bleeding. Prolonged bleeding episodes can be associated with trauma or may occur after invasive procedures, such as tooth extractions or tonsillectomies. True menorrhagia (> 80 mL of blood loss per cycle) occurs in 50-75% of affected women.3 Previously undiagnosed vWD may be present in up to 20% of women who have menorrhagia.21
In its severe form (type 3), bleeding characteristic of both primary and secondary hemostatic (fibrin clot formation) defects can be seen as a result of circulating factor VIII levels of 5% or less.17 In these cases, a defect in secondary hemostasis may lead to bleeding into soft tissues or muscles, or may result in a hemarthrosis similar to that found in patients with hemophilia.
Various conditions have been associated with increased vWF levels and may affect the initial diagnosis or course of the disease. These conditions include high estrogen levels, certain inflammatory states, advancing age, exercise, adrenergic stimuli, and pregnancy.17 In pregnancy, factor VIII:vWF levels rise, and peak in the third trimester.6,13,17,22 Shortly after delivery, these levels rapidly fall and increase the risk of bleeding in the early postpartum period.13,17,23
Hemophilia. Hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) are inherited bleeding disorders that are clinically indistinguishable. The severity of the disorder correlates well with the baseline factor activity level.8,9,18,24 Severe disease (factor activity level < 1%) occurs in about 70% of patients with hemophilia A, and about 50% with hemophilia B.9,12 In patients with severe disease, the onset of symptoms often occurs within the first year of life. In these individuals, a frequent recurrence of bleeding into joints and soft tissues commonly are encountered.8,12 Although it is rare even in severe disease, intracranial hemorrhage can occur at birth. Risk factors include a prolonged labor, the use of a vacuum extraction or forceps, or a precipitous delivery.8,12 Excessive bleeding following circumcision occurs in fewer than one-half of all severely affected males.8 Bleeding manifestations in patients with moderate disease may not be noted until the child is 1-2 years of age. In toddlers with moderate to severe disease, injuries requiring therapy often accompany the onset of walking and running. Moderate disease (factor activity level 1-5%) occurs in about 15% of patients with hemophilia A, and about 30% with hemophilia B.9,12 Mild disease (factor activity level > 5%) occurs in about 15% of patients with hemophilia A, and about 20% with hemophilia B.9,12 Episodes of spontaneous hemorrhage are rare in patients with mild disease, but significant bleeding still can be associated with major trauma or surgery.
Hemarthrosis is, by far, the most common and the most potentially debilitating manifestation of hemophilia. Bleeding can occur spontaneously in cases of moderate to severe disease, or may occur as a result of trivial trauma. The accumulation of blood within the joint space results in synovial proliferation and an increase in vascularity that predisposes the joint to further bleeding.8,24,25 Over time, the end result of multiple episodes of intra-articular hemorrhage can lead to a chronically deformed and painful joint. The knee most commonly is affected, but bleeding can occur in other joints, including the ankles, hips, wrists, shoulder, and fingers. Many hemophiliacs will experience a sensation of warmth and tingling prior to the onset of pain or swelling. This premonitory complaint may herald the presentation of an acute hemarthrosis. An examination of the affected joint may reveal a limited range of motion, swelling, tenderness, warmth, and surrounding muscular spasm. Prophylactic administration of factor replacement early in life has been employed successfully as a primary prevention strategy to limit the progression to hemophilic arthropathy.12
Another common manifestation of hemophilia is the development of subcutaneous and intramuscular hematomas.25 Although rarely life threatening, intramuscular hematomas account for almost one-third of all bleeding episodes in patients with hemophilia.8 Bleeding sites commonly include the forearm, calf, and thigh.24 Intramuscular hematomas often are limited by fascial planes and can lead to a compartment syndrome if vascular structures are compressed. Localized pain is the most common complaint when bleeding occurs into an extremity. When bleeding is retroperitoneal or involves the psoas muscle, presenting symptoms may include acute abdominal pain that mimics appendicitis, or numbness of the medial thigh from compression of the femoral nerve.5,8,24,25 The diagnosis of retroperitoneal bleeding can be confirmed by computed tomography. Minor abrasions, small hematomas, and the removal of sutures often do not require replacement therapy.
In patients with hemophilia, primary hemostasis may be temporarily effective in controlling hemorrhage after minor surgery or trauma. Delayed bleeding can occur hours or even days later. This can be a serious concern in a patient with a minor head injury, or when certain procedures such as tooth extractions are performed on an outpatient basis. Central nervous system (CNS) bleeding remains a major cause of death in the hemophiliac population, with a mortality rate approaching 30%.8 All patients reporting even minor head trauma should receive aggressive factor replacement because the complications associated with intracranial hemorrhage can be devastating. Intracranial hemorrhage has been reported to occur without preceding trauma; patients with persistent, unexplained headaches may require factor replacement.25 Emergency medicine physicians should have a low threshold for obtaining CT scans in patients at risk for an intracranial hemorrhage.
GI and genitourinary (GU) bleeding are other, less common manifestations of hemophilia. GI hemorrhage typically occurs as a result of an anatomic lesion proximal to the ligament of Treitz.8 The management of atraumatic GU bleeding ranges from reassurance and increased oral fluid intake for painless hematuria, to an extensive diagnostic workup and factor replacement.24,25
Sometimes referred to as hemophilia C, factor XI deficiency is inherited as an autosomal recessive trait.9,18 Although, the incidence of this disorder appears to be very rare in the general population (1 in 1,000,000 people), the prevalence may be as high as 4% in Ashkenazi Jews. The majority of the reported cases have been in Israel, Los Angeles, and New York.1,26 The clinical manifestations of this disorder, although variable, often are more mild than those encountered with cases of classic hemophilia. On occasion, these patients may remain clinically silent until challenged by surgery or trauma. Presently, no licensed factor XI concentrates are available in the United States. If needed, these patients can be treated with fresh frozen plasma (FFP).
Other Rare Coagulation Factor Deficiencies
Together, all other factor deficiencies are exceedingly rare and represent less than 1% of all factor deficiencies; all are inherited as autosomal recessive traits.1,26 Deficiencies of factor I (fibrinogen); factor II (prothrombin); and factors V, VII, and X can present with mucosal bleeding or easy bruising. Factor XIII deficiency may present with bleeding complications after trauma or surgery. In almost all cases of severe bleeding, these patients can be treated with an initial dose of 10 cc/kg of FFP.26 In patients with factor XIII or fibrinogen deficiency, cryoprecipitate (CPT) can be administered.
General Management of Bleeding Disorders
Comprehensive care is a mainstay of the long-term management of patients with hemophilia and other bleeding disorders. The National Hemophilia Program was initiated in 1975 and presently consists of 26 major hemophilia treatment centers and 124 affiliate centers.13 A multidisciplinary team approach is utilized to provide optimal patient care. When a patient presents to the ED, rapid triage, evaluation, and initiation of treatment should be employed to limit the hemorrhagic complications. When managing acute bleeding episodes, the emergency medicine physician should routinely enlist the patient’s hematologist or referral center to assist in further coordination of care.
The recommendation that patients should avoid using aspirin or other medications with antiplatelet effects is common to all bleeding disorders.23,27 The use of these medications may potentiate an underlying bleeding disorder, or may precipitate an episode of bleeding in a patient with otherwise occult disease. Acetaminophen with or without the addition of codeine is a reasonable alternative to the non-steroidal anti-inflammatory medications, which might otherwise be used for pain management. Accident prevention and avoiding contact sports also are important considerations in severe cases.
Routine venipuncture can be performed in a patient with a congenital bleeding disorder. After the procedure is performed, direct pressure should be applied for 3-5 minutes. Immunization schedules should be followed, and when injections are required they should be administered subcutaneously to avoid developing an intramuscular hematoma. After the injection, direct pressure should be applied to the site for five minutes.5
Treatment of von Willibrand’s Disease
To effectively manage an acute bleeding episode in a patient with vWD, it often is helpful to have knowledge of the particular disease subtype, and the patient’s prior response to currently available therapies. Specific management will depend on the location and extent of hemorrhage and the severity of the underlying disease. The mainstay of treatment for patients with vWD includes the use of transfusional (factor VIII replacement), non-transfusional desmopressin acetate [DDAVP]), and other adjunctive therapies.
Factor replacement with plasma-derived blood products often is required for patients who have contraindications, or who do not respond, to DDAVP, or who have severe or life-threatening hemorrhage. Presently, standard formulas do not exist for determining the specific amount of factor replacement needed to obtain hemostasis. Treatment often is empiric, and in the emergency department, efficacy generally is determined by the clinical response of the patient. It is recommended to raise factor VIII levels to at least 50%.17,27 As with hemophilia, the clinician should assume that during an acute bleeding episode the starting factor activity level is 0%. Raising factor VIII levels to within 30-50% of normal are sufficient to control most minor bleeding episodes; levels of 50-100% often are necessary to control limb- or life-threatening hemorrhage.12 These levels may need to be maintained for a number of days (usually 3-10), depending on the severity of the underlying disease and the location and extent of hemorrhage. In addition to replacing factor VIII to obtain adequate hemostasis, some authors recommend correcting a prolonged bleeding time.1,27 The clinical significance of a prolonged bleeding time as a predictor of hemorrhage during surgery still is unclear, and the absolute need to correct a prolonged bleeding time to obtain surgical hemostasis is yet unresolved.18 Many patients have successfully undergone a variety of surgical procedures without excessive blood loss after replacement of factor VIII and without correction of the bleeding time.4,23,27
In years past, FFP and CPT have been relied upon as the therapeutic agents of choice for the treatment of patients with vWD.27 FFP is the fluid portion of blood that is separated and frozen at 18°C within eight hours of collection, and contains plasma proteins including all coagulation factors. One unit of FFP contains about 7% of the coagulation factor activity of a 70 kg patient.28 CPT is prepared by thawing FFP between 1°C and 6°C and recovering the precipitate. CPT contains coagulation factor VIII, vWF (factor VIII:vWF complex), factor XIII, fibrinogen, and fibronectin. Each unit of CPT contains 80-100 IU of factor VIII:vWF, and has 5-10 times more vWF than FFP.17,23,28 CPT is considered a second-line agent for the management of vWD and hemophilia A, and should be used only if factor VIII concentrates are unavailable for the management of these patients.28 If CPT is used, a recommended starting dose is 2 bags/10 kg of body weight.23 This dose should raise the concentration of factor VIII to hemostatic levels. Additional therapy can be administered every 8-12 hours and adjusted based on laboratory parameters and the clinical response.23 FFP is not routinely used as a factor replacement for either vWD or hemophilia, and should be considered only if other agents, such as CPT and factor concentrates, are unavailable.13,28 Presently, donor blood screening for viral contamination includes anti-HIV, anti-HTLV-I/II, HIV-I-Ag, HbsAg, anti-HBc, anti-HCV, nucleic acid testing for HCV, and serology testing for syphillis.28 This testing has markedly reduced, but not eliminated the risk of viral transmission. Neither FFP nor CPT undergo routine viral inactivation. The use of these blood products still presents a very small but finite risk of viral transmission. Employing a limited pool of blood donors known not to be at risk for viral transmission to obtain CPT has been used successfully in the management of patients with vWD.27 Also, the yield of factor VIII:vWF in CPT can be increased two- to five-fold by the pretreatment of selected blood donors with DDAVP.13
Although early factor VIII concentrates revolutionized the treatment of hemophilia A, they were relatively ineffective for treatment of the mucocutaneous bleeding commonly associated with vWD because they contained little of the HMWMs of vWF.23 The role that vWF plays in primary hemostasis is largely dependent on the presence of these HMWMs in normal plasma concentrations.2 Commercial preparations of factor VIII that lack the HMWMs of vWF still may have a role in secondary hemostasis by binding to, and preventing, proteolytic degredation of factor VIII.2
Few of the available factor VIII concentrates can be used to effectively treat patients with vWF. (See Table 1.)
| Table 1. Factor VIII Concentrates Currently Available for the Treatment of Hemophilia A1,12,13,26 | |
| Intermediate and High-Purity Factor VIII Products | |
| Factor VIII SD (New York Blood Center) | |
| Humate-P (Behringwerke) | |
| Profilate OSD (Alpha) | |
| Melate (New York Blood Center) | |
| Koate HP (Bayer - Miles)—High purity | |
| Alphanate (Alpha)—High purity | |
| Hyate:C (Speywood/Porton)—Porcine plasma (For use in persons with factor VIII inhibitors) | |
| Monoclonal Antibody-purified Factor VIII Products | |
| Monoclate P (Armour) | |
| Hemofil M (Baxter - Hyland) | |
| AHF Method M (Red Cross) | |
| Recombinant Factor VIII Products—Synthetic | |
| Recombinate (Baxter) | |
| Bioclate (Centeon)—Identical to Recombinate | |
| Kogenate (Bayer - Miles) | |
| Helixate (Centeon)—Identical to Kogenate | |
Humate-P is an intermediate purity factor VIII concentrate that is available in the United States. This product contains a high level of HMWMs of vWF in a pattern that is similar to normal plasma.13,17,23,27 This product has been used successfully to treat many of the subtypes of vWD and has been efficacious in shortening bleeding times, correcting laboratory abnormalities, and managing acute bleeding episodes. After an infusion of factor VIII, most patients with vWD have an initial, predictable rise in factor VIII activity. Unlike patients with hemophilia A, with whom the half-life of the infused complex is about 12 hours, patients with vWD have a sustained but variable rise in the factor VIII activity that plateaus at about 24 hours but may persist for as long as 48-72 hours.9 In some patients, an initial fall of factor VIII activity is followed by a secondary delayed rise. This variable response to transfused factor VIII has been termed the secondary transfusion response and seems to be a feature unique to vWD.9 Factor VIII concentrates produced with purified monoclonal antibody and recombinant technology do not contain adequate levels of vWF and should not be used in the management of vWD.13,17 Specific guidelines for the treatment of vWD are listed in Table 2. Click here.
Treatment of Hemophilia
The cornerstone of the treatment of hemophilia is factor replacement. As with vWD, this is undertaken with the assumption that at the time of an acute hemorrhage, the factor activity level is 0%. An arbitrary division of the various bleeding manifestations of hemophilia into minor and major categories may prove useful for guiding initial factor replacement. For this purpose, a minor bleeding episode is one that is associated with an uncomplicated hemarthrosis, intramuscular or subcutaneous hematomas in noncritical areas, bleeding that occurs with minor dental procedures, or minor trauma.9 By definition, a major bleeding episode is a life-threatening or potentially life-threatening condition. These include hematomas in critical areas such as the neck and postoperative hemorrhage and intracranial hemorrhage. In managing acute bleeding episodes, the goal is to rapidly restore the coagulation factor activity to levels that provide adequate hemostasis. Although these values were determined empirically, the hemostatic level for factor VIII is approximately 25-30 u/dL (25-30%), and the hemostatic level for factor IX is approximately 15-30 u/dL (15-30%).8,9 Achieving these values should provide a plasma concentration of factor VIII or IX to control hemorrhage in most minor bleeding episodes. In more severe or life-threatening hemorrhage, factor replacement of 50-100% often is needed to provide adequate hemostasis.
The principles of initial factor replacement and maintenance dosing are based on the kinetics of in vivo recovery and survival of the factor infusion, and the hemostatic levels required to control the bleeding episode.9,12 Administration of 1 unit of factor VIII per kg of body weight will raise the plasma factor level by 2%.8,12,26 After an infusion, the initial recovery of factor VIII is approximately 80%, and its half-life in circulation is about 12 hours.9,12 Administration of 1 unit of factor IX per kg of body weight will raise the plasma factor level by 1%.8,12,26 After an infusion, the initial recovery of factor IX is approximately 25-50%, and its half-life in circulation is about 24 hours.9,12 Larger doses of factor IX are required to achieve the same plasma concentration because a greater amount of factor IX escapes into the extravascular space. If required, maintenance dosing of factor VIII and IX is administered at intervals of approximately every 12 hours and 24 hours, respectively.
All currently available factor concentrates in the United States are extremely safe with regard to the transmission of HIV and hepatitis. Vaccination in childhood against hepatitis can provide additional safety. Although rare, cases of hepatitis A and parvovirus B 19 (non-lipid-coated viruses) transmission have been reported with these products.1,8,26 Some products prepared using monoclonal antibody purification or recombinant technology contain trace amounts of murine protein; to date, no cases of transmission of animal viruses have been reported.26 Although recombinant factor VIII contains human albumin, no cases of HIV or hepatitis C transmission have been reported.12 Some concern has been raised that factor VIII concentrates of intermediate purity may suppress cellular immunity. This may play a role when choosing a specific factor replacement in patients with hemophilia A who are HIV-positive.1,26 Monoclonal antibody-purified, high-purity plasma-derived, or recombinant products seem to be reasonable choices for treating these patients. (See Tables 3 and 4. Table 3 appears below. To see table 4, click here.) Many authors recommend treating children with monoclonal antibody-purified or recombinant products for the added margin of viral safety.1,26
| Table 3. Factor IX Concentrates Currently Available for the Treatment of Hemophilia B1,12,13,26 | |
| Low-purity Factor IX Prothrombin Complex Concentrates | |
| Bebulin VH (Immuno) | |
| Profilnine (Alpha) | |
| Proplex T (Baxter - Hyland) | |
| Konyne 80 (Bayer - Miles) | |
| Activated Factor IX Prothrombin Complex Concentrates | |
| (For use in patients with factor VIII or IX inhibitors) | |
| Autoplex T (Baxter - Hyland) | |
| FEIBA VH (Immuno) | |
| Coagulation Factor IX Products | |
| High-purity factor IX | |
| — AlphaNine SD (Alpha) | |
| Monoclonal antibody-purified factor IX | |
| — Mononine (Centeon) | |
| Recombinant Factor IX—Synthetic | |
| Benefix (Genetics Institute) | |
Two types of factor IX replacement products currently are available for the management of hemophilia B, the prothrombin complex concentrates (PCCs), and the high-purity coagulation factor IX concentrates. In addition to factor IX, the PCCs contain prothrombin and factors VII and X, in a partially activated form. Frequent or prolonged administration of these products has been associated with the development of disseminated intravascular coagulation and other thrombogenic complications, including deep venous thrombosis, pulmonary embolism, and rarely, myocardial infarction.1,12,25 The high-purity factor IX concentrates contain little or no other coagulation proteins, and have little or no thrombogenic potential.
The development of alloantibodies to exogenously administered factor VIII or factor IX is a feared complication of hemophilia therapy. These IgG antibodies neutralize or inhibit factor replacement, making transfusion therapy less effective or ineffective. This condition occurs more often in persons with moderate-to-severe disease and has a prevalence of up to 15% in hemophilia A, and up to 3% in hemophilia B.1,25 Inhibitor antibodies may be identified on routine comprehensive screening or may be suspected when a bleeding episode does not respond to traditional factor replacement. Through plasma assays, patients are designated as either "low responders" (£ 10 Bethesda units/mL), or "high responders" (³ 10 Bethesda units/mL). Patients that are "low responders" usually do not develop an anamnestic response to factor VIII or factor IX replacement therapy. Bleeding in these individuals often can be controlled with higher doses (3-4 times the usual dose) and more frequent infusions of factor replacement.1,8,12,26 Patients classified as "high responders" to factor VIII or IX often are difficult to manage because no single therapy is uniformly successful.8 Bleeding episodes are refractory even to high-dose factor replacement. In persons with hemophilia A, a therapeutic option may be the administration of porcine factor VIII (Hyate:C) at a dose of 100-150 u/kg.12 As a result of species specificity, antibodies against human factor VIII are less likely to inhibit the effectiveness of porcine factor VIII. PCCs (100 units/kg) or activated PCCs (50-75 u/kg) also can be used in the treatment of both minor and major bleeding episodes in persons with hemophilia A or hemophilia B with high responding inhibitors. These complex concentrates act by bypassing the requirement for factor VIII or IX in clot formation.12 Recombinant factor VIIa also has been used successfully to treat patients who have acquired "high responding" antibodies to factor VIII.12,22
In adults, factor concentrate should be administered by slow intravenous push at a rate not to exceed 3 mL/min, and a rate not to exceed 100 u/min in young children.5 Specific guidelines for the treatment of hemophilia are listed in Table 4.
Adjunctive Agents for Managing Bleeding Disorders
Antifibrinolytic agents such as epsilon-aminocaproic acid and tranexamic acid often are used in combination with DDAVP or factor replacement therapy. By inhibiting plasminogen, these medications help to prevent clot lysis in persons with hemophilia or vWD who are undergoing dental procedures or tonsillectomies or who have oral cavity bleeding.17,26 Epsilon-aminocaproic acid is administered orally in a dose of 6 g every six hours for adults and 100 mg/kg for children. An intravenous preparation is available for patients who cannot swallow. Tranexamic acid is administered orally in a dose of 25 mg/kg every six hours (maximum, 1.5 g), or 10 mg/kg (maximum, 1.0 g) intravenously every eight hours.9,12,25 It is not recommended to use these agents for the treatment of hemarthrosis or bleeding within the GU tract. In addition, these agents are not to be used in patients with hemophilia B who are concomitantly receiving PCCs because of the increased the risk of thrombosis.5
Estrogen therapy can increase factor VIII:vWF levels and may be useful for women with vWD who have chronic menorrhagia. Other agents, such as gelfoam, topical thrombin, and microfibrillar collagen, may be helpful in selected cases.
Non-Transfusional Therapy. DDAVP (1-deamino-8-D-arginine vasopressin), a synthetic derivative of antidiuretic hormone (vasopressin), promotes hemostasis by causing a release of factor VIII:vWF from cellular storage sites. DDAVP has been shown to be an effective hemostatic agent (increasing levels of factor VIII:vWF by 3-5 times), and an alternative or adjunctive therapy to factor replacement in many patients with vWD and mild hemophilia A.29-34 When administered intravenously (0.3 mcg/kg over 20 minutes), desmopressin can be highly effective in managing minor bleeding episodes. A concentrated nasal spray (300 mcg in adults [2 sprays] or 150 mcg in children £ 50 kg [1 spray]) also has been efficacious in controlling bleeding.34,35 Subcutaneous (0.3 mg/kg) or intranasal administration has allowed for home therapy in selected cases. DDAVP also has been effectively used as a prophylactic agent in preparation for elective surgical procedures in patients with mild hemophilia A and vWD.29,31,33 If used as a prophylactic agent to prevent surgical bleeding, it is suggested that an infusion trial be performed to determine efficacy prior to surgery.
DDAVP infusion may be associated with a few mild side effects, including facial flushing, a transient headache, or fatigue.32,33 Hyponatremia and seizures have occurred when used in children younger than 2 years of age.38
Most patients with type 1 vWD and mild hemophilia A have an excellent response to DDAVP.34,36,37 A less consistent, but generally positive hemostatic effect occurs in patients with type 2N and type 2M vWD.34,36 Patients with type 2A vWD often will have a variable or poor response, and some patients with type 2B vWD have developed thrombocytopenia after the administration of DDAVP.34,36,37 Patients with type 3 vWD, severe hemophilia A, or hemophilia B do not respond to DDAVP.
Summary
Although not encountered on a daily basis, patients with congenital bleeding disorders will have hemorrhagic complications that require emergency care. As emergency medicine physicians, we often are faced with the responsibility of initiating this care without the luxury of bedside specialty consultation. Being knowledgeable about the various diseases and treatment options available will help to expedite the management of these patients and improve their clinical outcome.
References
1. Cohen AJ, Kessler CM. Treatment of inherited coagulation disorders. Am J Med 1995;99:675-682.
2. Schneppenheim R, Thomas KB, Sutor AH. Von Willebrand disease in children. Sem Thromb Hemost 1995;21:261-275.
3. Nichols WC, Ginsburg D. von Willebrand disease. Medicine (Baltimore) 1997;76:1-20.
4. Werner EJ. von Willebrand disease in children and adolescents. Ped Clin North Am 1996;43:683-707.
5. Hemophilia of Georgia. Protocols for the Treatment of Hemophilia and von Willebrand Disease. Available at www.wfh.org. Accessed on March 28, 2001.
6. Bloom AL. von Willebrand factor: Clinical features of inherited and acquired disorders. Mayo Clin Proc 1991;66:743-751.
7. Polycythemia. In: Cotran RS, Kumar V, Tucker C, et al, eds. Robbins Pathologic Basis of Disease. 6th ed. W. B. Saunders Co; 1999.
8. Kessler CM. Coagulation Factor Deficiencies. In: Russell CL, Bennett JC, Goldman L, eds. Cecil Textbook of Medicine. 21st ed. W. B. Saunders Co.; 2000.
9. Rodgers GM, Greenberg CS. Inherited Bleeding Disorders. In: Lee RG, Foerster J, Lukens J, et al, eds. Wintrobe’s Clinical Hematology. 10th ed. Lippincott Williams & Wilkins, Inc; 1999.
10. Hamilton G, Janz TG. Disorders of Hemostasis. In: Rosen P, Barkin R, eds. Emergency Medicine: Concepts and Clinical Practice. 4th ed. St. Louis, MO: Mosby-Year Book Inc.; 1998.
11. Soucie JM. Occurrence of hemophilia in the United States. Am J Hematol 1998;59:288-294.
12. DiMichele D, Neufeld EJ. Hemophilia. Hematol Oncol Clin North Am 1998;12:1315-1344.
13. Hemophilia Program Fact Sheet, updated 4/99. Available at www.mchb.hrsa.gov.
14. Parker RI. Coagulation. In: Civetta JM, Taylor RW, Kirby RR, eds. Critical Care, 3rd ed. Lippincott-Raven Publishers; 1997.
15. Hemostasis and Thrombosis. In: Cotran RS, Kumar V, Tucker C, et al, eds. Robbins Pathologic Basis of Disease. 6th ed. W. B. Saunders Co.; 1999.
16. Schafer AI. Approach to the Patient with Bleeding and Thrombosis. In: Russell CL, Bennett JC, Goldman L, eds. Cecil Textbook of Medicine, 21st ed. W. B. Saunders Co.; 2000.
17. Rick ME. Diagnosis and Management of Von Willebrand’s Syndrome. Med Clin North Am 1994;78:609-623.
18. Basic Tests in Hematologic Disorders. In: Ravel R, ed. Clinical Laboratory Medicine. 6th ed. St. Louis, MO: Mosby-Year Book Inc.; 1995.
19. Sadler JE. A Revised classification of von Willebrand disease. Sem Thromb Hemost 1994;71:520-525.
20. Ewenstein BM. The pathophysiology of bleeding disorders presenting as abnormal uterine bleeding. Am J Obstet Gynecol 1996;175: 770-777.
21. Edlund M, Blomback M, von Schoultz B, et al. On the value of menorrhagia as a predictor for coagulation disorders. Am J Hematol 1996;53:234-238.
22. Lusher JM. Screening and diagnosis of coagulation disorders. Am J Obstet Gynecol 1996;175:778-780.
23. Logan LJ. Treatment of von Willebrand’s disease. Hematol Oncol Clin North Am 1992;6:1079-1094.
24. Lusher JM, Warrier I. Hemophilia A. Hematol Oncol Clin North Am 1992;6:1021-1033.
25. Pfaff JA, Geninatti M. Hemophilia. Emerg Med Clin North Am 1993;11:337-363.
26. Lusher JM. Transfusion therapy in congenital coagulopathies. Hematol Oncol Clin North Am 1994;8:1167-1180.
27. Aledort LM. Treatment of von Willebrand’s disease. Mayo Clin Proc 1991;66:841-846.
28. Circular of Information for the Use of Human Blood and Blood Components. American Red Cross; Revised January 1999.
29. Mannucci PM, Ruggeri ZM, Pareti FI, et al. DDAVP: A new pharmacological approach to the management of haemophilia and von Willebrand’s disease. Lancet 1977;1:869-872.
30. Mannucci PM, Canciani MT, Rota L, et al. Response of factor VIII/von Willebrand factor to DDAVP in healthy subjects and patients with hemophilia A and von Willebrand’s disease. Br J Haematol 1981;47:283-293.
31. Warrier AI, Lusher JM. DDAVP: A useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228-233.
32. Kobrinsky NL, Gerrard JM, Watson CM, et al. Shortening of bleeding time by 1-deamino-8-D-arginine vasopressin in various bleeding disorders. Lancet 1984;1:1145-1148.
33. de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willibrand’s disease to treatment with desmopressin. Ann Intern Med 1985;103:6-14.
34. Sutor AH. Desmopressin (DDAVP) in bleeding disorders of childhood. Sem Thromb Hemost 1998;24:555-566.
35. Seremetts SV, Aledort LM. Desmopressin nasal spray for hemophilia A and type 1 von Willebrand disease. Ann Intern Med 1997;126: 744-745.
36. Sutor AH. DDAVP is not a panacea for children with bleeding disorders. Br J Haematol 2000;108:217-227.
37. Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders: The first 20 years. J Am Soc Hematol 1997;90: 2515-2521.
38. Smith TJ, Gill JC, Ambruso DR, et al. Hyponatremia and seizures in young children given DDAVP. Am J Hematol 1989;31:199-202.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.