Loss of Estrogen Receptor Alpha and its Contribution to Tumorigenesis in Breast Cancer
Loss of Estrogen Receptor Alpha and its Contribution to Tumorigenesis in Breast Cancer
By Christine Couldrey, PhD, and Jeffrey E. Green, PhD
Estrogens play a pivotal role in sexual development, reproduction, and physiological processes in a variety of tissues, including mammary, pituitary, bone, and liver tissue, as well as the cardiovascular system.1 However, estrogens also are involved in various pathological processes such as breast and endometrial cancer.2 At the present time, the primary treatment regimen for breast cancer involves endocrine therapies that inhibit estrogen signaling. One of the major challenges to improving the treatment of breast cancer is understanding and overcoming the resistance to endocrine therapy that often develops during the course of treatment.
Estrogen elicits its effect through the activation of estrogen receptors (ER). These receptors belong to a super family of nuclear hormone receptors that includes retinoic acid, progesterone, glucocorticoid, androgen, and thyroid hormone receptors. To date, two genes that encode ERs have been identified (ERa and ERb). These genes are expressed in a tissue-specific and temporal manner, and are influenced by estrogen and other hormones.3-5 ERa is the major form of ER in the mammary gland and in breast cancers; therefore, the following discussion will focus on the loss of ERa expression in breast cancer.
Molecular Mechanisms of Estrogen Receptor Expression
The ERa gene is made up of eight exons, encoding a protein of 595 amino acids that can be divided into six conserved functional domains. (See Figure 1.) Estradiol (the most potent form of estrogen) elicits its effect by binding to ER, causing a conformational change in the protein, displacement of heat shock proteins, and phosphorylation of the receptor. These changes allow receptor homodimerization and subsequent binding to cis-acting elements within enhancers of target genes (see Figure 2), such as c-myc, progesterone receptor, and TGF-a, ultimately leading to the proliferation of ER-positive cells.
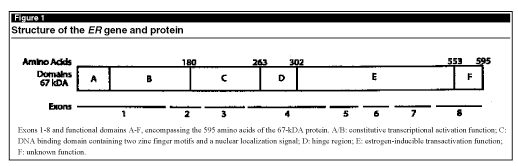 |
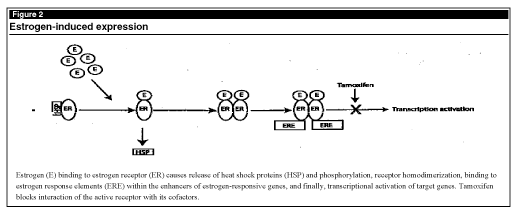 |
Approximately two-thirds of human breast cancers express ER, and their growth is stimulated by estrogen. For these tumors, therapeutic strategies include estrogen ablation or the administration of anti-estrogens (e.g., tamoxifen). The remaining one-third of breast cancers lack ER. These tumors can grow in the absence of estrogen, rarely respond to hormonal therapy, and are associated with less differentiated, more aggressive tumors and shorter disease-free survival.6 Given the importance of ER and the fact that all current therapies act through ER, defining how and why tumors become ER-negative is a critical step in improving therapeutic outcomes. Unfortunately, the molecular mechanisms underlying the lack of ER expression are poorly understood.
Loss of ERa During Tumorigenesis
A number of factors influencing ERa expression have been identified.7 Loss of functional ERa may occur because of aberrations in the ER gene (i.e., mutations or rearrangements), or because of changes at the transcriptional, post-transcriptional, or translational levels.
Genomic Alterations
The human ERa gene is located on chromosome 6q25. Although homozygous deletion containing this region of chromosome 6 has not been reported, loss of heterozygosity (LOH) has been noted in 80-90% of breast cancers. However, mutation of one allele and loss or replacement of a chromosomal segment on the other were not found to be accompanied by changes in ER expression.8
Point mutations that cause functional alterations of the ER protein also are rare. One study of 188 breast cancer patients that used single-stranded polymorphism analysis, denaturing gradient gel electrophoresis, and DNA sequencing reported that loss of ER was not the result of mutations in the coding region of the ERa gene.9 Together, these results suggest that genetic alterations in the ERa gene at the DNA level might account only for a small portion of hormone independence. However, it is important to note that breast tumors are a heterogenous population that includes normal and malignant cells; current technologies may not be sufficiently sensitive to identify ER mutations that occur infrequently.
Splicing Variants of ER mRNA
In contrast to the minimal number of mutations that have been identified, a significant number of ERa splicing variants have been found.10 Moreover, ERa splicing variants have been found to occur at a high frequency in breast carcinomas. Alternative splicing, resulting in deletion of one or more exons (2, 3, 4, 5, or 7), has been detected and the resulting biological properties have been examined. An in-depth description of all splicing variants is beyond the scope of this review; however, they can be categorized into three groups: constitutively active variants, dominant-negative variants, and inactive variants.8,10,11 However, the true in vivo, functional significance of these variants is complex, because the variants always are found in conjunction with wild type ERa mRNA and they frequently are detected in normal as well as malignant tissues.
As a further level of complexity, the human ERa gene has two major promoters. Each of these promoters yields unique transcripts that encode the same size gene product; both promoters are potentially active, although usage varies across cell and tissue types.8,10 To date, no firm correlations have been made between promoter usage and ERa loss in the progression of breast cancer.
Methylation of the ERa Gene
Given the small proportion of cancers with ER-negative phenotypes that can be explained by disruptions at the genomic level, other mechanisms must exist for the loss of ERa expression. Analysis of human breast cancers indicates that the loss of ERa protein is due to a lack of ERa mRNA.10 It is possible that reduced levels of ERa mRNA are the result of reduced levels of active transcription cofactors required for the transcription of this gene. However, to date, this possibility has not been widely demonstrated. Another, more commonly studied, epigenetic mechanism for transcriptional failure is DNA hypermethylation of the ERa gene.
The methylation of CpG islands of DNA induces a dilation of a major groove and a kink in a minor groove at the opposite side of the double helix loop. This conformational change in the chromosome alters the interaction between DNA and histone particles. DNA methylation in the promoter region of many genes is associated with transcriptional silencing of the gene either through a direct effect or via a change in the chromatin conformation that inhibits transcription.
The ERa gene has been studied as a target for silencing via methylation. Initial studies that focused on the methylation status of the body of the gene failed to show any correlation between methylation and ERa expression.8 However, more recent studies directed at the CpG island in the 5’ transcriptional regulatory region and first exon of the gene have established a clear correlation between ERa CpG island methylation and lack of ERa gene expression in breast cancer cell lines and primary breast tumors.12 A functional role for methylation is supported by studies in which two human ER-negative breast cancer cell lines were treated with demethylating agents (5-Aza-2’deoxycytidine or 2-Azacytidine), resulting in the demethylation of the ERa gene CpG islands, re-expression of the ERa gene, and production of ER.13 Together, these data suggest that ERa gene CpG island methylation may play a role in gene silencing in at least a subset of ER-negative human breast cancers. Al-though, given the complex nature of transcription, it is likely that other transcriptional regulators of ER also are involved in breast tumor progression.
ERa Degradation—mRNA and Protein
It is possible that reduction of ERa mRNA seen in ER-negative breast cancer is due to decreased mRNA stability and therefore, is a possible cause of the ER- negative phenotype. However, few studies have addressed this issue and data are not sufficient to draw any conclusions.
Determination of ERa status in normal and malignant tissue is most commonly performed by immunostaining for the protein. The absence of ER also may be due to enhanced degradation of the protein. However, given the previously described evidence showing that steady-state mRNA levels are severely reduced, control of ERa expression appears to be at the RNA levels. Thus, few studies have examined ERa protein degradation.
Use of Animal Models—Why Use Animal Models?
Although a significant amount of breast cancer research has been performed on human tissue, research using human tissues from biopsies and tumor removal has several severe limitations. First, the amount of excised tissue is limited, and must be used primarily for assessing tumor characteristics (prognosis) by a pathologist. Second, after biopsy/tumor removal from the patient, a significant length of time often elapses before the tumor can be stored in a manner such that DNA, protein, and particularly, RNA are not degraded. Furthermore, tissue treatment required by the pathologist often is not compatible with subsequent molecular analysis. Third, it is extremely difficult to obtain multiple samples from the same patient, making time course studies virtually impossible. Finally, the ethical issues involved and the difficulty of obtaining consent are complex when dealing with human tissue.
During the past 10 years, numerous mouse models of mammary cancer have been generated through chemical carcinogen, knockout, or transgenic technologies. These models have overcome the problems associated with the use of human tissue (i.e., significant quantities of tumors may be collected and tumor progression and histopathology are reproducible). However, other problems have arisen. The main question of current concern is how similar these tumors are to those seen in human breast cancers. While no single model can answer every question given the variety of human breast cancers seen today, certain models are well-suited to studying particular aspects of mammary tumorigenesis. The C3(1)/Tag model of mammary cancer has proven to be useful.
C3(1)/Tag Transgenic Mouse Model
All female mice, heterozygous or homozygous for the C3(1)/Tag transgene, develop mammary carcinomas.14 The transgene is expressed in mammary epithelial ductal cells without stimulations of pregnancy or artificial hormones. Expression that is not dependent on pregnancy is unlike many other transgenic mouse models of breast cancer that use hormone-responsive promoters to drive oncogenes and require one or more pregnancies to express the transgene. Pregnancy-induced tumorigenesis does not closely mimic breast cancer in humans, in whom pregnancy is a well-accepted protective factor. Furthermore, mammary lesions develop during a predictable time course, with many histological similarities to human ductal carcinoma in situ and infiltrating ductal adenocarcinomas.15
It has been shown recently that the C3(1)/Tag transgene is not estrogen-responsive. These studies also led to the discovery that ERa expression appears to be lost during mammary tumor progression, a detrimental prognostic factor in human breast cancer as described above. Loss of ERa in the C3(1)/Tag model appears to be the first demonstration of ERa loss in a transgenic mouse model, although it remains to be determined whether other models also display this phenotype. ERa was detected by immunohistochemistry and western blotting in normal mammary epithelial cells and low-grade mammary intraepithelial neoplasia (MIN).16 Tumors that were classified as high-grade MIN showed a reduction in ERa protein. ERa expression in invasive carcinomas was low to undetectable. Northern blot analysis of mammary gland and invasive carcinoma mRNA from these mice correlated with protein levels. Thus, it appears that ERa loss in mammary tumors from C3(1)/Tag mice is not the result of increased ERa protein degradation, but rather a result of a decrease in steady-state ERa mRNA, either through a reduction in stability or reduced transcription. Given the literature linking ERa gene hypermethylation with decreased ERa transcription in breast cancer and prostate cells lines,12,17 it is possible that hypermethylation also is the cause of ERa loss in C3(1)/Tag tumors. Thus, given the clinical implications of ERa loss outlined above, this may be an extremely useful model for studying the poorly understood molecular mechanisms of ERa loss.
Although the mouse is used extensively as a model organism to study ERa regulation in vivo, the complete structure of the ERa gene has not been elucidated. Given the complex structure of the human gene with two alternative promoters and numerous mRNA variants, it is possible that the mouse ERa gene may exhibit similar features. Studies recently published and studies in progress indicate that the mouse ERa gene is a complex transcription unit with tissue-specific promoter usage and alternate splicing.18 This complexity, along with gaps in our knowledge of ERa structure, has made it difficult to study methylation in the mouse ERa gene.
ERa Expression—A Potential Target for Breast Cancer Treatment?
The discussion presented above illustrates the desperate need for a greater understanding of why a significant proportion of human breast cancers stop expressing ERa and subsequently become refractory to treatment. The use of mouse models of breast cancer enables studies of tumor development and progression, and the development of new therapies for breast cancer treatment that prevent the loss of ERa, or cause re-expression of the gene so that endocrine therapies remain/become effective in halting tumor progression. (Dr. Couldrey is Postdoctoral Fellow, and Dr. Green is Head, Transgenic Oncogenesis Group, Laboratory of Cell Regulation and Carcinogenesis, National Cancer Institute, NIH, Bethesda, MD.)
References
1. George FW, Wilson JD. Sex determination and differentiation. In: Knobil E, et al, eds. The Physiology of Reproduction. New York: Raven; 1988:3-26.
2. Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer: The Richard and Hinda Rosenthal Foundation award lecture. Cancer Res 1988;48: 246-253.
3. Koritnik DR, Koshy A, Hoversland RC. 17beta-estradiol treatment increases the levels of estrogen receptor and its mRNA in male rat liver. Steroids 1995;60: 519-529.
4. Saceda M, Lippman ME, Chambon P, et al. Regulation of the estrogen receptor in MCF-7 cells by estradiol. Mol Endocrinol 1988;2:1157-1162.
5. Shupnik MA, Gordon MS, Chin WW. Tissue-specific regulation of rat estrogen receptor mRNAs. Mol Endocrinol 1989;3:660-665.
6. Samaan NA, Buzdar AU, Aldinger KA, et al. Estrogen receptor: A prognostic factor in breast cancer. Cancer 1981;47:554-560.
7. Green S, Walter P, Kumar V, et al. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986;320:134-139.
8. Ferguson AT, Lapidus RG, Davidson NE. The regulation of estrogen receptor expression and function in human breast cancer. In: Foon KA, Muss HB, eds. Cancer Treatment and Research. Boston: Kluwer Academic Publishers; 1998:255-278.
9. Roodi N, Bailey LR, Kao WY, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst 1995;87:446-451.
10. Iwase H, Omoto Y, Iwata H, et al. Genetic and epigenetic alterations of the estrogen receptor gene and hormone independence in human breast cancer. Oncology 1998;55(suppl 1):11-16.
11. Fasco MJ. Estrogen receptor mRNA splice variants produced from the distal and proximal promoter transcripts. Mol Cell Endocrinol 1998;138:51-59.
12. Iwase H, Omoto Y, Iwata H, et al. DNA methylation analysis at distal and proximal promoter regions of the oestrogen receptor gene in breast cancers. Br J Cancer 1999;80:1982-1986.
13. Ferguson AT, Lapidus RG, Baylin SB, et al. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res 1995;55: 2279-2283.
14. Maroulakou IG, Anver M, Garrett L, et al. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A 1994;91: 11236-11240.
15. Green JE, Shibata MA, Yoshidome K, et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: Ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 2000; 19:1020-1027.
16. Yoshidome K, Shibata MA, Couldrey C, et al. Estrogen promotes tumor development in C3(1)/SV40 large T-antigen transgenic mice: Paradoxical loss of estrogen receptor alpha expression during tumor progression. Cancer Res 2000;60:6901-6910.
17. Li LC, Chui RM, Nakajima K, et al. Frequent methylation of estrogen receptor in prostate cancer: Correlation with tumor progression. Cancer Res 2000;60: 702-706.
18. Kos M, O’Brien S, Flouriot G, et al. Tissue-specific expression of multiple mRNA variants of the mouse estrogen receptor alpha gene. FEBS Lett 2000;477: 15-20.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.