Special Feature: Rhabdomyolysis
Rhabdomyolysis
By William J. Brady, MD
Rhabdomyolysis is defined as injury to skeletal muscle resulting in breakdown of muscle tissue with subsequent leakage of intracellular contents, including myoglobin, potassium and other electrolytes, creatine phosphokinase (CPK), and other muscle enzymes. These same intracellular elements serve as diagnostic markers for the syndrome. Rhabdomyolysis classically involves the triad of muscle weakness, myalgias, and darkened urine. As is true with most such traditional constellations of symptoms and signs, this classic triad is noted on presentation in few patients. More commonly, the patient complains of muscular aches and weakness, prompting the diagnosis of a benign viral syndrome. The term myoglobinuria—incorrectly used as a synonym for rhabdomyolysis—indicates only the presence of myoglobin in the urine and, therefore, is a less descriptive term for the clinical syndrome; it is best avoided in clinical practice.
Etiology and Pathophysiology
The potential causes of rhabdomyolysis are numerous, including exposure to ethanol, various medications and other toxins, trauma (e.g., crush injuries and orthopedic fractures), prolonged compression of limbs, excessive seizure activity, electrolyte disorders, strenuous activities (e.g., military basic training, weight lifting, and marathon running), and muscle cellular enzyme deficiency states. Ethanol abuse and its sequelae, limb compression, and prolonged seizure activity were the three most commonly encountered etiologic factors of rhabdomyolysis in a series of hospitalized patients.1 A significant number of cases, however, still are classified as "idiopathic" despite detailed histologic, enzymatic, and biochemical studies.
The pathophysiology of rhabdomyolysis must be understood on cellular (microscopic) and muscle compartment (macroscopic) levels. The final common pathway in the cellular pathophysiology of rhabdomyolysis, regardless of etiology, is believed to be an impairment of either production or utilization of adenosine triphosphate (ATP). The reduction in ATP, whether relative or absolute, causes energy-requiring reactions and homeostatic mechanisms to fail. The macroscopic pathophysiology of rhabdomyolysis involves local hydrostatic and osmotic pressure conditions within the myofascial compartment. When the intracompartmental pressure in-creases above a specific level due to such factors as hemorrhage, inflammatory fluid, or external compression, perfusion to the compartment is impaired, resulting in disruption of the skeletal muscle metabolic processes. The net effect is increased intracompartmental pressure and further disruption of perfusion to the closed space of the myofascial compartment as well as to distal structures of that vascular distribution.
Clinical Presentation
The presentation of rhabdomyolysis ranges from very straightforward to very elusive. Patients may complain only of myalgias and weakness. Alternatively, the patient may present as a multiple trauma victim with a compartment syndrome. Examination may reveal localized or diffuse muscular tenderness, focal muscular weakness, edema, skin changes consistent with pressure necrosis, or findings of a compartment syndrome.
The laboratory diagnosis of rhabdomyolysis includes both urine and serum examinations. A urine specimen with a dipstick positive for blood and simultaneously demonstrating zero-to-few red blood cells is consistent with rhabdomyolysis, due to the presence of myoglobin. While serum and urine myoglobin levels can confirm these findings, they are not reliable indicators of rhabdomyolysis due to rapid plasma clearance; the serum half-life of myoglobin is only 1-3 hours and there is poor correlation of myoglobinuria with myoglobinemia. (See Figure.)2,3 CPK levels have been shown to be the most sensitive marker of myocyte injury and rhabdomyolysis.4
Figure:
Release Kinetics of Serum Markers in Rhabdomyolysis
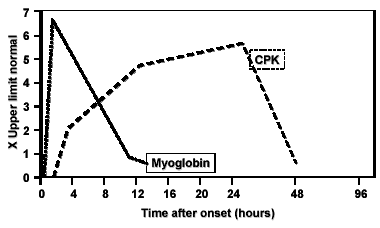
The use of CPK measurement is the most appropriate laboratory method for confirming the diagnosis of rhabdomyolysis due to a number of factors, including the ease of test performance in most clinical settings, near-immediate appearance of measurable levels of CPK in the serum after muscle injury, and lack of rapid clearance of CPK from the blood. (See Figure.) CPK levels (usually 100% MM fraction) at least five times above the upper limit of normal are required to fulfill the criteria for rhabdomyolysis.5 The absolute height of the CPK elevation, however, does not translate to disease severity and risk of complication. The CPK level peaks at 24-36 hours after the skeletal muscle "insult" and declines at a rate of approximately 40% per 24 hours. Failure of CPK levels to decline at the appropriate rate suggests an ongoing process of skeletal muscle injury.
Electrolyte abnormalities also are seen in rhabdomyolysis. Hyperkalemia, resulting from excessive release of potassium from damaged muscle, renal failure, and metabolic acidosis, can appear within the first few days after the onset of the illness. Hypocalcemia is thought to occur from the deposition of calcium salts in damaged skeletal muscle. Hypercalcemia also has been noted during the diuretic phase of myoglobin-induced renal failure, when serum phosphate concentrations fall and the calcium salts are reabsorbed. Hyperphosphatemia results from phosphate release during muscle injury. Hyperuricemia, which tends to be much higher in patients with exertional rhabdomyolysis, results from purines released from injured muscle cells. Concentrations of blood urea nitrogen and creatinine in the serum may increase, resulting from both acute renal failure and release from skeletal muscle.
Complications
Complications of rhabdomyolysis include various electrolyte abnormalities, acute renal failure (ARF), compartment syndrome, disseminated intravascular coagulation, acute respiratory failure (rare), and cardiomyopathy (very rare). The most feared complication of rhabdomyolysis is renal failure due to acute tubular necrosis. Renal injury results from myoglobin casts obstructing renal tubules, a decreased glomerular filtration rate, and direct nephrotoxicity of ferrihemate, a breakdown component of myoglobin. Furthermore, hypovolemia and any process producing urinary acidification predispose to acute renal failure. With urine pH less than 5.6, myoglobin dissociates into ferrihemate and a globin moiety. Ferrihemate is a dose-related nephrotoxin. The toxic effect of ferrihemate on a cellular level has been shown to be a result of ferrihemate-related production of free hydroxy radicals. Researchers at Denver General Hospital developed the discriminant function equation to predict the risk of ARF in such patients. The equation is as follows: R = 0.7 (potassium) + 1.1 (creatinine) + 0.6 (albumin) 6.6. The R values and the associated risk of ARF are noted: R < 0.1 (0% chance of ARF) and R > 0.1 (41% chance of ARF).1
Treatment
Treatment of patients with rhabdomyolysis includes not only an attempt at identification of the triggering event but also management of metabolic complications and organ dysfunction. The clinician must make a diligent search for any reversible cause of rhabdomyolysis (e.g., compartment syndrome) and halt ongoing muscle damage. Saline (0.9%) loading by intravenous route is the mainstay of therapy in that it restores intravascular volume and induces a solute diuresis. All patients should have a urinary catheter to adequately monitor fluid output. Diuresis can be accomplished using an osmotic agent (mannitol) or a loop diuretic (furosemide). Sodium bicarbonate may protect the kidneys from the effects of myoglobinuria by rapidly increasing urinary pH, provided that the development of frank metabolic alkalosis is avoided.6 The use of sodium bicarbonate is based upon the theoretical advantage of inhibiting the formation of the nephrotoxin ferrihemate. Hyperkalemia is treated in the usual manner with infusion of insulin, glucose, and calcium gluconate, inhalation of a nebulized beta-agonist agent, administration of oral and rectal exchange resins, electrocardiographic monitoring, and elimination of potassium intake. Dialysis may be required to correct electrolyte abnormalities or to treat oliguric renal failure. Finally, hospital admission is required to treat the syndrome with the aim of halting progression to renal failure, monitoring for other associated complications, and ensuring that the process of skeletal muscle death is not ongoing or does not recur.
Dr. Brady, Associate Professor of Emergency Medicine and Internal Medicine, Vice Chair, Emergency Medicine University of Virginia, Charlottesville, is on the Editorial Board of Emergency Medicine Alert.
References
1. Gabow PA, et al. The spectrum of rhabdomyolysis. Medicine 1982;61:141-152.
2. Koskelo P, et al. Kinetic behavior of 131I-labeled myoglobin in human beings. Clin Chem Acta 1967;17: 339-347.
3. Stone MJ, et al. Radioimmunoassay of myoglobin in human serum. Results in patients with acute myocardial infarction. J Clin Invest 1975;56:1334-1339.
4. Hess JW, et al. Serum creatinine phosphokinase (CPK) activity in disorders of heart and skeletal muscle. Ann Intern Med 1964;61:1015-1028.
5. Knochel JP. Rhabdomyolysis and myoglobinuria. Semin Nephrol 1981;1:75-86.
6. Ron D, et al. Prevention of acute renal failure in traumatic rhabdomyolysis. Arch Intern Med 1984;144:277-280.
Rhabdomyolysis is defined as injury to skeletal muscle resulting in breakdown of muscle tissue with subsequent leakage of intracellular contents, including myoglobin, potassium and other electrolytes, creatine phosphokinase, and other muscle enzymes.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.