Common Electrolyte Problems in Pediatric Patients Presenting to the ED
Common Electrolyte Problems in Pediatric Patients Presenting to the ED
Authors: Ronald M. Perkin, MD, MA, FAAP, FCCM, Professor and Chairman, Department of Pediatrics, Brody School of Medicine at East Carolina University; Medical Director, University Health Systems of Eastern Carolina Children’s Hospital, Greenville, NC; William E. Novotny, MD, Associate Professor of Pediatrics, Pediatric Critical Care, Brody School of Medicine at East Carolina University; Glenn D. Harris, MD, Associate Professor of Pediatrics, Director, Pediatric Diabetology, Brody School of Medicine at East Carolina University; and Irma Fiordalisi, MD, Professor of Pediatrics, Pediatric Critical Care, Director, Pediatric Intensive Care Unit, Brody School of Medicine at East Carolina University.
Peer Reviewer: Robert Danoff, DO, MS, Family Practice Residency Program Director, Frankford Hospitals, Philadelphia, PA.
Electrolyte disorders are an important sequelae of many diseases that result in pediatric visits to general and pediatric emergency departments (EDs). Accurate identification of such abnormalities has the potential to impact treatment, outcome, and disposition of these patients.
Electrolyte panels are a common tool used to evaluate children who present to EDs. Although this test is relatively inexpensive on an individual basis, studies have shown that ordering serum electrolyte tests regularly can be costly.1 It has been estimated that in excess of $1 billion per year could be saved in U.S. health care costs with small modifications to clinicians’ patterns of ordering electrolytes.1
While sets of high-yield criteria for obtaining electrolytes have been identified in the adult and elderly ED populations, few studies have identified criteria for obtaining this test in children. One study found that the presence of any of the following criteria identified all children with clinically significant electrolyte abnormalities: age younger than 6 months, vomiting, delayed capillary refill, dry mucous membranes, tachycardia, or the presence of diabetes mellitus.1 The patient population in this study was primarily well children with acute illness. Further analysis will be required to determine whether these or other predictors might aid overall electrolyte panel decision making.
Changes in volume and composition of body fluids due to disorders of fluid and electrolyte balance cause or result from various common clinical illnesses. Following introductory comments about body fluid volume and composition, an overview of some of the etiologies of the disorders of volume, tonicity, and composition of body fluids and the therapy to correct these disorders is provided. The focus is on abnormalities of cation concentration: sodium, potassium, calcium, and magnesium.— The Editor
Sodium, Osmolality, and the Volume of Body Fluids
Total body water, which is 55-72% of body mass, varies with sex, age, and fat content, and is distributed between the intracellular and extracellular spaces.2 The extracellular fluid (ECF), which comprises about one-third of total body water, includes the intravascular plasma fluid and the extravascular interstitial fluid. Plasma ions include primarily sodium (Na+), chloride (Cl-), and bicarbonate (HCO3-), which are excluded from intracellular environments, and lesser amounts of potassium (K+), magnesium, calcium, phosphates, sulfates, organic acids, and protein. Interstitial fluid, which surrounds the cells, has the same composition as plasma, but with less protein. The principal components of intracellular fluid (ICF) are potassium, proteins, magnesium, sulfates, and phosphates.
In the ECF, Na+ and Cl- constitute 90% or more of the effective solutes. Serum Na+ concentration defines the relative amount of sodium and water in plasma; the maintenance of a normal Na+ concentration, thus, contributes to regulation of the volume of body fluids.2 The size of the ECF and ICF compartments depends on the amount of water within each; the distribution of water depends on their osmolality. It is important to recognize that osmolality and tonicity are not identical. Osmolality is a measure of the number of solute particles per unit volume. Measured osmolality of a solution includes all particles whether they are osmotically active or inactive. Sodium chloride or other impermeable solutes such as mannitol or glucose do not readily cross cell membranes to enter cells. They remain restricted to the ECF space and are thus "effective osmols" by obligating water to remain extracellularly. Cell membranes are freely permeable to solutes such as urea, methanol, or ethanol. While contributing to measured osmolality, these "ineffective osmols" do not obligate water to remain in the ECF space. Tonicity takes into account only osmotically active impermeable solutes and is the important physiological parameter.
In a given patient, the osmolality may be calculated as follows, using the values of 2.8 and 18 to convert values of blood urea nitrogen (BUN) and glucose, respectively, to mOsm/L:
Osmolality = 2[Na+ in mEq/L] + [BUN in mg/dL]/2.8 + [glucose in mg/dL]/18.
Normal serum osmolality is maintained by kidney function, which dilutes or concentrates urine. This is accomplished by a variety of mechanisms involving glomerular filtration; arterial pressure; blood flow; physical factors in the kidneys; the sympathetic nervous system; and hormones such as aldosterone, atrial natriuretic factor, vasopressin, and dopamine.2 These systems converge to control water and electrolyte balance through glomerular ultrafiltration of the plasma followed by changes in the electrolyte content of this ultrafiltrate by tubular reabsorption and secretion. These mechanisms, together with thirst, control both plasma osmolality and plasma volume.
Hyponatremia
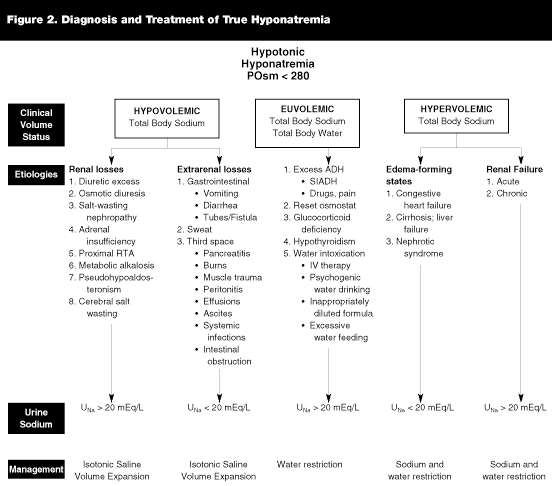
Hyponatremia is generally defined as a plasma sodium concentration of less than 130 mEq/L (130 mmol/L). When assessing the child with hyponatremia, it is important to determine whether the low serum sodium is true or fictitious. It also is important to determine the patient’s volume status (i.e., euvolemia, hypovolemia, or hypervolemia). This allows the basic distinction between hyponatremia caused by sodium loss or hyponatremia caused by an increase in total body water (TBW), resulting in a relative dilution of the ECF compartment. The serum sodium concentration generally cannot be used to estimate the patient’s total body fluid status. Figures 1 and 2 summarize the causes of hyponatremia.
 |
 |
The severity of signs and symptoms of hyponatremia is dependent on the rapidity of its development and the degree of decline in plasma osmolality.3-5 As the plasma osmolality decreases, an osmotic gradient across the blood-brain barrier develops, which results in water movement into the brain. This cerebral overhydration is responsible for the majority of symptoms seen in hyponatremia. In general, neurologic manifestations do not occur until the serum sodium concentration is less than 125 mEq/L. However, patients with preexisting neuropathology may exhibit symptoms at higher serum sodium levels. Signs and symptoms may include headache, agitation, disorientation, lethargy, nausea, vomiting, muscular cramps, decreased deep tendon reflexes, seizures, coma, pseudobulbar palsies, and signs of cerebral herniation.3,6 The rapid development of hyponatremia (< 24 hours) carries significant morbidity and mortality.3,5-7
Within 24 hours, the brain cells partially adapt to the overhydration or hypoosmolality by gradually losing intracellular osmotically active solutes. Along with these solutes, water leaves the brain cells and partially reverses the cerebral edema. Therefore, patients whose hyponatremia developed over several days or weeks are less symptomatic than patients whose hyponatremia developed acutely.3 These concepts are important to remember when correcting hyponatremia.
Hyponatremia is a common cause of seizures in infants; it is the leading cause of afebrile seizures in this population.8-13 It is frequently due to water intoxication.10 Child abuse should be considered as part of the differential diagnosis of children who present with hyponatremic seizures.9,13
Hyponatremic seizures usually are generalized, with frequent progression to status epilepticus.10 Respiratory failure requiring intubation is not uncommon, although most require mechanical ventilation for fewer than 24 hours. Decreased body temperature, another finding in acute symptomatic hyponatremia, occurs in 80-90% of hyponatremic patients.10 Hyponatremic seizures are more difficult to treat than simple febrile seizures.8 Correction of the underlying electrolyte disturbance is more effective than the use of anticonvulsants.10 It is now well established that prompt and appropriate therapy with adequate amounts of sodium can lead to recovery in children with hyponatremic encephalopathy, even after respiratory arrest.9,14
Hyponatremia occurs as a result of water retention, or sodium loss, or both; therefore, the ECF volume may be low, normal, or high. An analysis of the urine sodium concentration may be a diagnostic aid in determining the etiology of the hyponatremia.
Since serum sodium concentration is the main determinant of plasma osmolality, hyponatremia usually reflects hypo-osmolality.15,16 Initially, one must determine whether hyponatremia reflects true hypotonicity. There are two clinical circumstances in which hyponatremia may be present without hypotonicity. High levels of plasma lipids or proteins increase plasma volume, which decreases the percentage of plasma volume that is water. Because many clinical laboratories measure and report sodium concentrations in mEq/L of plasma (not in mEq/L of plasma water), the reported sodium concentration will be artificially low. Newer methods of sodium measurement that use ion selective electrodes measure serum sodium activity directly in plasma water and are not affected by the proportion of serum occupied by lipids and proteins. A clinical clue to such "pseudohyponatremia" is the presence of lipemic serum and a normal measured serum osmolality (because lipids or proteins have large molecular weights and, therefore, are minor components of overall plasma osmolality). In patients with nephrotic syndrome, a clinical rule of thumb is that an increase of triglycerides of 1 g/dL will decrease serum sodium concentrations by 2 mEq/L.15
The second situation in which a patient may be hyponatremic, but not hypotonic, occurs when an osmotically active solute (e.g., glucose or mannitol) has been added to the extracellular space. Water drawn out of cells by such an osmotically active particle will dilute the serum sodium concentration despite isotonicity or even hypertonicity. When hyperglycemia is present, a clinically useful method for bedside prediction of the serum sodium concentration once normoglycemia is achieved is that the serum sodium concentration is lowered by 1.6 mEq/L for each 100 mg/dL increase in glucose concentration.16 The presence of an osmotically active nonglucose solute in the extracellular space is identified readily by comparing the calculated osmolality with measured osmolality. If they differ by more than 10 mOsm/L, it can be inferred that some other solute (mannitol or glycerol) may be present.16 Such an osmolal gap might be the first clue to ingestion of a foreign substance such as methanol or ethanol.
Hyponatremia with Hypovolemia. Most instances of hyponatremia presenting in the ED are caused by volume depletion that is the result of an underlying disease; thus, appropriate fluid administration, as well as treatment of the underlying disease, usually is required to correct the hyponatremic state.4 Extrarenal losses most commonly occur via the gastrointestinal tract (vomiting or diarrhea) but may be secondary to other disorders such as burns, pancreatitis, or peritonitis. The normal kidney responds to these hypovolemic states by conserving sodium and water; thus, a hypertonic urine with a sodium concentration of less than 20 mEq/L should be expected. In hyponatremic hypovolemic patients with urine sodium concentrations greater than 20 mEq/L, the possibility of adrenal insufficiency exists, particularly if the serum potassium concentration is elevated. The most common cause in the infant and young child is the salt-losing form of congenital adrenal hyperplasia. Other causes include congenital adrenal hypoplasia, acute infection, hemorrhage into the adrenal glands, inadequate replacement of adrenocorticosteroids, and inappropriate tapering of steroids.
Hyponatremia is a well-recognized complication of traumatic brain injury and subarachnoid hemorrhage and frequently is ascribed to the syndrome of inappropriate antidiuretic hormone secretion (SIADH) or to cerebral salt wasting (CSW). The latter, which first was described in 1950, is characterized by hyponatremia, diuresis, and natriuresis in the face of a contracted ECF, in contrast to SIADH, in which patients are euvolemic.17-22 The distinction is important, as CSW is thought to be caused by excess secretion of natriuretic peptide and the treatment is saline replacement, whereas fluid restriction would be the treatment of SIADH.
The treatment of hyponatremia in the volume-depleted patient should be directed toward expansion of the ECF volume with salt-containing solutions. Replacement with salt-containing solutions will correct systemic and renal hemodynamics and allow the normal function of the osmoreceptor-antidiuretic hormone system to normalize plasma osmolality. Enough fluid should be given to provide maintenance requirements plus the volume of deficit. The adequacy of volume repletion must be evaluated from the findings on physical examination and laboratory data. Attention must be directed to the underlying disease and attempts made to control abnormal ongoing losses. When shock is present, isotonic saline can be given rapidly using 10-20 mL per kg intravenously over 10-20 minutes. This can be done repeatedly until systemic arterial blood pressure is restored and urine produced.
In addition to adequate water and salt, patients with adrenal insufficiency will require efforts to correct hypoglycemia and hyperkalemia, and will need replacement of deficient adrenal corticosteroids.23 If a patient presents with a salt-losing crisis, high doses of hydrocortisone (50-140 mg/m2 per day in 3-4 divided doses) are required.23
Acute symptomatic hyponatremia is a medical emergency for which the use of hypertonic saline may be required.24-27 Because of the dangers of congestive heart failure, osmotic demyelination syndrome, or cerebral hemorrhage, only enough hypertonic saline should be used to correct serum sodium to levels of 125 mEq/L. The amount of hypertonic saline needed can be calculated using the following formula:
mEq Na+ = (0.6) (body wt in kg) (125 [Na+]),
where [Na+] = serum sodium concentration in mEq/L.
In the water intoxicated patient, spontaneous water diuresis occurs so rapidly that treatment with hypertonic saline is usually unneccessary.14
Neurologic complications as a result of overly rapid correction of hyponatremia have been reported in adults and older children.3,5 The syndrome of osmotic demyelination, or central pontine myelinolysis, is the result of neuronal dehydration. Hyponatremia-induced losses of intracellular potassium and amino acids shift the cells to a hypotonic state, predisposing them to acute dehydration when exposed to a normotonic or hypertonic solution aimed at rapidly correcting hyponatremia. Caution is required when contemplating rapid correction of acute symptomatic hyponatremia.3
Euvolemic Hyponatremia. In patients with hyponatremia who have neither contraction of extracellular fluid volume nor expansion to the point of clinical edema, SIADH should be considered. Patients with SIADH have a concentrated urine in spite of the hyponatremia, and the urinary sodium concentration closely parallels the intake and is usually greater than 20 mEq/L.
There are several factors that affect antidiuretic hormone (ADH) secretion, the most important of which is plasma osmolality.3 Hyponatremia from SIADH occurs due to water retention secondary to a persistently elevated ADH level inappropriate to any osmotic stimuli that normally affects ADH secretion.
SIADH is seen in a variety of clinical disorders that can be subdivided into four categories: increased hypothalamic production of antidiuretic hormone because of disease or the action of drugs; ectopic production of antidiuretic hormone; potentiation of antidiuretic hormone as an effect of drugs; and exogenous administration of antidiuretic hormone. The diagnosis ultimately rests on the demonstration of inappropriately high plasma concentration of arginine vasopressin when hyponatremia has appeared spontaneously or has been induced by an increase of water intake. In the absence of levels, the following criteria can be used: 1) hyponatremia with corresponding plasma hypo-osmolality; 2) inappropriately elevated urine osmolality relative to plasma osmolality; 3) normal renal function; 4) normal adrenal and thyroid functions (particularly ruling out glucocorticoid deficiency and hypothyroidism); 5) high urine sodium excretion in the presence of normovolemia; 6) absence of clinical signs of hypovolemia and dehydration; 7) absence of edema-forming states or evidence of volume depletion; and 8) correction of hyponatremia and natriuresis by fluid restriction.15 SIADH commonly is seen with disorders that affect the central nervous system, in the presence of pulmonary diseases, and with use of certain drugs.
The management of SIADH can be considered under two headings: treatment of the hyponatremia and treatment of the disease process responsible for the syndrome. Since all the signs of uncomplicated SIADH result from excessive retention of water, they all respond to simple restriction of fluids.3 Reduction of fluid intake to the point where urinary and insensible losses induce a negative water balance will lead to restoration of normal body fluid volume, a reduction in urinary sodium excretion, and an increase in serum sodium concentration. Ordinarily, a fluid intake of 50-75 percent of maintenance will accomplish this goal. Full restoration of serum sodium concentration requires that some sodium be made available. However, excessive amounts of sodium are promptly excreted in the urine. In some cases, the hyponatremia is so severe or has occurred so rapidly that signs of water intoxication (convulsions, coma) develop. In such instances, a rapid form of treatment is preferred. The recommended treatment is hypertonic saline and furosemide.15, 27 This combination will cause a rise in serum sodium concentration and concurrent water diuresis. This treatment is reserved for emergency situations and should be followed by fluid restriction.
It must be stressed that in the presence of high vasopressin levels (appropriate or inappropriate) the imprudent administration of hypotonic fluids can culminate in severe cerebral edema.7,17,27
Hyponatremia with Hypervolemia (Dilutional Syndromes). Hypervolemic hyponatremia occurs when the net water retention exceeds that of sodium. Clinically, it may be seen with: 1) edema-forming states such as congestive heart failure, cirrhosis, and nephrosis; and 2) acute or chronic renal failure. Under normal conditions, an increase in sodium intake will result in sodium and water retention and an increase in intravascular volume. This increase in intravascular volume will in turn result in an increase in renal perfusion and in subsequent activation of the afferent and efferent mechanisms controlling renal sodium excretion. The net effect is an increase in renal sodium excretion in an attempt to return the intravascular volume to normal.2 In the presence of disease states such as congestive heart failure, nephrotic syndrome, and cirrhosis, afferent and efferent mechanisms of sodium retention are activated.
These patients have an increase in total body sodium but even larger increases in total body water. Therapy should be directed at maximal improvement of the underlying disorder. Since total body sodium already is elevated, efforts to increase serum sodium through administration of saline will result only in further expansion of ECF volume and may worsen the clinical status of the patient. Attempting to decrease total body water by severe restriction of fluid is the most appropriate therapy. The excess sodium needs to be treated concomitantly by sodium restriction and judicious use of diuretics.
Hypernatremia
Hypernatremia, defined as a rise in the serum sodium concentration to a value exceeding 145 mmol/L, is a common electrolyte disorder.28 Because sodium is a functionally impermeable solute, it contributes to tonicity and induces the movement of water across cell membranes.28,29 Therefore, hypernatremia invariably denotes hypertonic hyperosmolality and always causes cellular dehydration, at least transiently. The resultant morbidity may be inconsequential, serious, or even life-threatening.
Hypernatremia represents a deficit of water in relation to the body’s sodium stores, which can result from a net water loss or a hypertonic sodium gain. Net water loss accounts for the majority of cases of hypernatremia.3,15,28 It can occur in the absence of a sodium deficit (pure water loss) or in its presence (hypotonic fluid loss). Hypertonic sodium gain usually results from clinical interventions or accidental sodium loading. Intentional salt poisoning also has been described as a form of child abuse.30
Because sustained hypernatremia can occur only when thirst or access to water is impaired, the groups at highest risk are patients with altered mental status, infants, and elderly patients. Hypernatremia in infants usually results from diarrhea.
Clinical signs and symptoms of hypernatremia with associated hypertonicity are directly related to cerebral cell dehydration resulting from water movement from the ICF to the ECF. Neurologic manifestations include varying degrees of depressed sensorium, ranging from lethargy to coma.3,28 The majority of patients exhibit marked irritability, a high-pitched cry, and seizure activity. Muscle tone may be normal or increased, and may be accompanied by hyperreflexia or twitching. Examination of the cerebrospinal fluid may show an elevated protein without pleocytosis. Some patients may also have hyperglycemia, hypercalcemia or hypokalemia and/or metabolic acidosis.3 With extreme hypertonicity and resultant water movement from brain cells into ECF, the entire brain can shrink away from the cranium and produce rupture of cerebral vessels. Consequently, focal intracerebral and subdural hemorrhages and venous thrombosis may occur.15,28
Since the central nervous system particularly is vulnerable to hypertonicity, the brain cells adapt within hours by increasing intracellular osmolality with resultant movement of water back into the brain cells. Myoinositol, taurine, glycerylphosphorylcholine, and betaine are the organic osmolytes (formerly called idiogenic osmoles) responsible for normalizing brain water content.29 If hypertonicity develops rapidly, these intracellular osmoles or osmoprotective molecules may not be generated fast enough to prevent brain cell shrinkage. Therefore, the severity of clinical manifestations of hypernatremia and associated hypertonicity is relative to its degree and rate of development.
The mechanism of hypernatremia with normal volume status is excessive sodium intake. The majority of cases are iatrogenic, including the use of sodium bicarbonate or improperly diluted infant formula. Ultimately, even these disturbances will result in a deficit of water since water will be lost as the kidney attempts to excrete the salt load.
In clinical medicine, the most common cause of hypernatremia is primary water deficit in excess of sodium, resulting in hypernatremia with decreased volume status.28,31 Water and sodium loss may occur via the extrarenal route or the renal route. Included in this category are patients with diabetes insipidus (DI). DI is characterized by complete or partial failure of ADH secretion (central DI) or renal response to ADH (nephrogenic DI), resulting in excretion of hypotonic urine.31 Central DI may be idiopathic, but the majority of affected patients have a history of head trauma, central nervous system infections, or tumors. Nephrogenic DI may be congenital or acquired, and results in hypernatremia usually associated with decreased water intake.
Hypernatremic dehydration also is well described in breastfed newborns.32 The serum sodium concentration in these cases may be elevated dramatically and frequently is associated with complications. These infants may present in the first week of life with fever, and the absence of overt signs of dehydration. Serum bilirubin concentrations also may be elevated. Within a few hours of rehydration, the fever subsides quickly, and the serum bilirubin concentrations fall rapidly. In general the infants make an uneventful recovery without permanent neurological sequelae. Such cases emphasize the importance of early and regular measurement of body weight in exclusively breast-fed infants.
Hypervolemic hypernatremia results when sodium gain is greater than water intake. This may include administration of hypertonic saline or sodium bicarbonate. Hypernatremia with increased volume status also may be seen in patients with primary hyperaldosteronism or Cushing’s syndrome because of sodium retention.
Whenever possible, therapy of hypernatremic patients should be directed at the underlying disease process (e.g., administration of vasopressin analogues in DI). In the presence of shock or severe ECF volume contraction, restoration of the intravascular volume takes precedence over normalization of plasma osmolality. In this setting, isotonic saline solutions are the recommended fluid replacement to restore circulating blood volume.
The speed of correction of hypernatremia depends on the rate of its development and the accompanying clinical presentation. Correction of hypernatremia should be accomplished slowly, except in the setting of acute massive salt poisoning. Rapid lowering of serum sodium may result in water movement from the ECF into the brain cells, resulting in cerebral edema and possible herniation. When serum sodium acutely exceeds 175 mEq/L (as in salt poisoning), dialysis may be performed to rapidly lower serum sodium. Once serum sodium concentration is at 170 mEq/L, further reduction should be carried out over 48 hours, with the aim to lower serum sodium at a rate no greater than 1 mEq/L/hour.28
A slower pace of correction is prudent in patients with hypernatremia of longer or unknown duration, because the full dissipation of accumulated brain solutes occurs over a period of several days. In such patients, reducing the serum sodium concentration at a maximal rate of 0.5 mEq/L/hr prevents cerebral edema and convulsions.28
Hyperkalemia
Potassium is the major intracellular cation; only a very small fraction of total body potassium is in the intravascular space.33 Increased potassium concentration in serum is infrequent in pediatrics, but it can be life-threatening because of its effect on membrane potentials, particularly of heart muscle.
The serum potassium concentration is affected primarily by the kidney. Potassium is filtered by the glomerulus, then reabsorbed and secreted by the tubule. Processes that interfere with filtration or secretion (e.g., acute or chronic glomerulonephritis, interstitial nephritis) may cause hyperkalemia; processes that interfere with reabsorption may cause hypokalemia.33,34
The most common cause of an increased serum potassium is "pseudohyperkalemia" due to hemolysis or to tissue hypoxia distal to the placement of a tourniquet at the time the specimen was obtained. A repeat determination generally is sufficient to resolve whether hyperkalemia is present. True hyperkalemia in children most commonly is associated with acute or chronic renal failure. Certain drugs, used infrequently in pediatric practice, can produce hyperkalemia: spironolactone, amiloride, triamterene, cyclosporin, and angiotensin-converting enzyme inhibitors (e.g., captopril, enalopril).33,34 Overuse of nonsteroidal anti-inflammatory agents may produce hyperkalemia, secondary to chronic renal injury.
Clinical signs of hyperkalemia generally are absent until the serum potassium is quite high, particularly when the accumulation of potassium has occurred gradually. At concentrations higher than 7 mEq/L (generally higher than 8.5 mEq/L), the patient may develop ascending muscle weakness to the point of flaccid paralysis. Cerebration and cranial nerve function are preserved. There generally are no clinical signs of altered cardiac function until concentrations are higher than 9 or 10 mEq/L, when the patient may develop ventricular fibrillation or asystole.
Changes in the wave form on electrocardiography provide clues to the degree of hyperkalemia.35,36 The earliest changes are tall, peaked T waves, with a normal or shortened QT interval and shortened PR interval; these are present at serum potassium concentrations of 5.5 to 6.5 mEq/L. As the serum potassium concentration rises, cardiac conduction is impaired. The QRS complex begins to widen and the PR interval to lengthen at serum potassium concentrations of 6.5 to 7.5 mEq/L. With further increases in the serum potassium concentration, the P waves become broad and of low amplitude, the QT interval is prolonged, and the ST segment is either elevated or depressed (7.0 to 8.0 mEq/L). When the serum potassium concentration exceeds 8.0 mEq/L, P waves disappear; the QRS becomes markedly widened and may progress to a "sine wave" configuration, identifying the patient to be at risk of ventricular fibrillation or asystole.
Potential treatment (see Table 1) includes: 1) protecting the heart from the effects of hyperkalemia (e.g., calcium ion); 2) shifting potassium from the intravascular space to the intracellular space (e.g., sodium bicarbonate, insulin/glucose); and 3) eliminating potassium from the body (e.g., dialysis, ion exchange resins).
| Table 1. Emergency Treatment of Hyperkalemia Onset/Duration | ||||
| Technique | Agent | Dose | Onset/Duration of Action | Comment |
| Reversal of membrane effects | 10% Calcium gluconate | 0.5 mL/kg | Min/30-60 min | ECG monitor; discontinue if pulse rate < 100. |
| Movement of K into cells | Sodium bicarbonate, 7.5% (1 mEq = 1 mL) | 2-3 mL/kg | 30 min/1-4 hr | May use in the absence of acidosis. |
| Glucose 50% plus insulin (regular) | 1 unit for every 5-6g glucose | Same | Monitor blood glucose. | |
| Albuterol 0.5% solution | 0.01-0.05 mL/kg (max 1 mL) aerosolized with 1-2 ml saline | 15-30 min | Monitor heart rate. | |
| Enhanced excretion of K | Kayexalate | 1 g/kg | Hours/variable | Can be given PO or by rectum. Give with sorbitol (70%) |
| Key: IV = intravenous; ECG = electrocardiogram; K = potassium; PO = orally. |
||||
|
|
||||
An intravenous infusion of calcium ion acts rapidly but its effects last only approximately 1 hour.33,34 Sodium bicarbonate or insulin/glucose cause potassium to move intracellularly. Sodium bicarbonate has no significant action on plasma potassium in the first 60 minutes after administration.37-39 Insulin binds to specific membrane receptors and via a second messenger, stimulates the sodium-potassium adenosine triphosphatase pump resulting in intracellular uptake of potassium.37 This effect is independent of its hypoglycemic action. In children, a glucose load of 0.5g/kg/hr should be given. This is because many children will increase their endogenous insulin production with the administration of a glucose load. If blood glucose is elevated, insulin can be added starting at 0.05u/kg/hr.37
ß2-agonists drive K+ into cells by increasing Na-K-ATPase activity. They are effective in reducing the serum potassium concentration rapidly and can be delivered by aerosol.40 Studies have shown that albuterol in some patients can lower the K+ by up to 1.5 mEq/L within 30 minutes.39,41
Effects of calcium, insulin, bicarbonate, and ß2-agonists are transient. Therefore, these measures usually are followed by a cation exchange resin, diuretics, or dialysis to remove the excess K+ from the body. Sodium polystyrene sulfonate resin (Kayexalate) is given at a dose of 1 g/kg in 20% sorbitol orally or in a 70% sorbitol rectal installation. The latter must be retained for 20-30 minutes and can be repeated every 4-6 hours. In the gut, this resin takes up K+ and, to lesser degrees, Ca+2 and Mg+2, while it releases Na+. Each gram of resin may bind 1 mEq of K+ and release 1-2 mEq of Na+. The major side effects are nausea, constipation, hypokalemia, and retention of Na+ exchanged for K+. Loop and thiazide diuretics increase urinary K+ excretion by enhancing K+ secretion in the distal nephron but are not widely used because patients with impaired renal function are unlikely to respond. In acute or chronic renal failure, the above measures are used while preparations are made for dialysis. Hemodialysis is preferred to peritoneal dialysis because the rate of K+ removal is faster in stable patients with acute renal failure.
Hypokalemia
Hypokalemia is defined as a serum potassium level less than 3.5 mEq/L. As with hyperkalemia, nerves and muscles (including the heart) are most affected by hypokalemia, particularly if the patient has other, preexisting disease, such as cardiovascular disease.33,41
Both the total body stores of potassium and its distribution within the body are closely regulated by key hormones. The normal transcellular distribution of potassium (a high ratio of intracellular to extracellular potassium) is maintained by at least two hormonal signals that promote the entry of this cation into cells. Both insulin and b-adrenergic catecholamines increase cellular potassium uptake by stimulating cell-membrane Na+/K+ -ATPase.42 For insulin, there is a feedback system in which hyperkalemia stimulates insulin secretion and hypokalemia inhibits it.41 No feedback system has been identified for b-adrenergic stimulation, but b-blockade increases serum potassium and b-agonists decrease it, an effect that is independent of body stores of potassium. Synthesis of Na+/K+ ATPase is also stimulated by thyroid hormone, which may contribute to the hypokalemia that occurs in patients with hyperthyroidism.41 Administration of alkali causes a shift of potassium into cells, but the response is quite variable. In general, serum K+ decreases by approximately 0.3 mEq/L for every 0.1 increase in pH above normal.36 In patients with end-stage renal disease, administration of bicarbonate has only a slight effect on the transcellular distribution of potassium.43
It remains unclear whether aldosterone affects the transcellular distribution of potassium, but this hormone is clearly the major regulator of body stores of potassium through its effect on the excretion of potassium by the kidney.44 As in the case of insulin, there is a feedback control; hyperkalemia stimulates the release of aldosterone (with synergy from angiotensin II), and hypokalemia inhibits it. Other hormonal and nonhormonal factors modulate renal potassium excretion, but they do not appear to have a role in normal potassium homeostasis.
The regulation of extracellular potassium concentration and body stores of potassium is asymmetric. Depletion of potassium and hypokalemia can occur simply through a reduction in potassium intake and can persist for long periods of time, despite normal hormone signaling and renal function.41 Hyperkalemia, by contrast, elicits a brisk response and only is sustained when there is continued disruption or impairment of the normal regulatory systems.
Patients with hypokalemia often have no symptoms, particularly when the disorder is mild (serum potassium, 3.0-3.5 mEq/L). With more severe hypokalemia, nonspecific symptoms, such as generalized weakness, lassitude, and constipation, are more common. When serum potassium decreases to less than 2.5 mEq/L, muscle necrosis can occur, and at serum concentrations of less than 2.0 mEq/L, an ascending paralysis can develop, with eventual impairment of respiratory function.41,45 The likelihood of symptoms appears to correlate with the rapidity of the decrease in serum potassium. In patients without underlying heart disease, abnormalities in cardiac conduction are extremely unusual, even when the serum potassium concentration is below 3.0 mEq/L. In patients with cardiac ischemia, heart failure, or left ventricular hypertrophy, however, even mild-to-moderate hypokalemia increases the likelihood of cardiac arrhythmias.41 Hypokalemia increases the arrhythmogenic potential of digoxin. Thus, hypokalemia should be avoided or treated promptly in patients receiving digitalis derivatives.
Hypokalemia is rarely suspected on the basis of clinical presentation; the diagnosis is made by measurement of serum potassium. A low serum potassium concentration indicates disruption of normal homeostasis, with one very rare exception. In some patients with leukemia and markedly elevated white cell counts, potassium is taken up by the abnormal cells if the blood is left at room temperature for several hours.46 More commonly, hypokalemia in patients with leukemia is the result of renal potassium wasting.
Hypokalemia is suggested by changes in the ECG, including:
- U waves;
- T wave flattening;
- ST segment changes;
- Arrhythmias (especially if the patient is taking digoxin); and
- Pulseless electrical activity (PEA) or asystole.
Hypokalemia almost always is the result of potassium depletion induced by abnormal losses of potassium. More rarely, hypokalemia occurs because of an abrupt shift of potassium from the extracellular compartment into cells. In either case, drugs prescribed by physicians are the most common causes of hypokalemia. Thus, the first step in the management of hypokalemia is to review the patient’s drug record.
In the absence of an inciting drug, hypokalemia can result from an acute shift of potassium from the extracellular compartment to cells, from inadequate intake, or from abnormal losses. Most commonly, hypokalemia is the result of either abnormal loss through the kidney induced by metabolic alkalosis or loss in the stool induced by diarrhea. A brief description of the more common etiologies of hypokalemia follows.
b2-Sympathomimetic Drugs. A wide range of drugs have b2-sympathomimetic activity, including decongestants and bronchodilators. A standard dose of nebulized albuterol reduces serum potassium by 0.2 to 0.4 mEq/L, and a second dose taken within one hour reduces it by almost 1 mEq/L.41,47 The hypokalemia caused by these drugs is sustained for up to four hours.
Diuretics. The most common cause of hypokalemia is diuretic therapy. Both the thiazide and loop diuretics block chloride-associated sodium reabsorption (with each inhibiting a different membrane-transport protein) and, as a result, increase delivery of sodium to the collecting tubules, where its reabsorption creates a favorable electrochemical gradient for potassium secretion.41 The degree of hypokalemia is directly related to the dose of the thiazide diuretic. The combined use of furosemide or bumetanide with metolazone invariably causes moderate-to-severe hypokalemia, despite potassium supplementation. Diuretic-induced hypokalemia is usually but not always associated with a mild-to-moderate metabolic alkalosis (serum bicarbonate concentration, 28-36 mmol per liter). The diuretic drug acetazolamide, however, promotes potassium excretion by impeding hydrogen-linked sodium reabsorption and thus causes a metabolic acidosis along with hypokalemia.
Losses in Stool. The concentration of potassium in stool is 80-90 mEq/L, but because of the low volume of water in normal stool, only about 10 mmol usually is lost each day. In diarrheal states, the potassium concentration in stool decreases, but large quantities of potassium can nonetheless be lost as the volume of stool increases. Anything that increases stool volume, from infectious diarrhea to chemotherapy, can result in clinically significant potassium depletion and hypokalemia.39,41
Loss Through the Kidney. Large amounts of potassium are lost through the kidney in patients with a variety of disorders. For ease of diagnosis, these disorders are categorized according to acid-base status.
Metabolic Alkalosis. Hypokalemia is an almost invariable consequence of metabolic alkalosis. In the most common form of this disorder, induced by selective chloride depletion due to vomiting or nasogastric drainage, hypokalemia develops during the induction of alkalosis as a result of increased renal potassium loss. In the chloride-sensitive form of metabolic alkalosis, the administration of chloride corrects the alkalosis and allows the repletion of body stores of potassium if potassium intake is adequate.
More rarely, metabolic alkalosis occurs independently of chloride depletion as a result of systemic or intrarenal abnormalities that augment sodium reabsorption in the distal nephron. The most common of these abnormalities is primary hyperaldosteronism, a disorder often heralded by severe hypokalemia (serum potassium, < 3.0 mEq/L). Hypokalemia also may develop in patients with Cushing’s syndrome, but it usually is milder than in patients with hyperaldosteronism.41
Genetic abnormalities that influence the activity of renal ion transporters are rare causes of metabolic alkalosis and hypokalemia.41,48 Two of these disorders (Liddle’s syndrome and 11b-hydroxysteroid dehydrogenase deficiency) stimulate reabsorption of sodium by collecting duct cells and cause the syndrome of apparent mineralocorticoid excess, so named because this transport abnormality results in hypertension and hypokalemia, but serum aldosterone concentrations are low rather than high. In two other disorders, genetic mutations inactivate or impede the activity of chloride-associated sodium transporters in the loop of Henle (Bartter’s syndrome) and early distal tubule (Gitelman’s syndrome), causing metabolic alkalosis and hypokalemia without hypertension.48
Metabolic Acidosis. Hypokalemia is a cardinal feature of type I or classic distal renal tubular acidosis. The degree of hypokalemia in this disorder is not directly correlated to the degree of acidosis but more likely reflects dietary sodium and potassium intake and serum aldosterone concentrations. Life-threatening hypokalemia (serum potassium, < 2.0 mEq/L) may occur in patients with untreated distal renal tubular acidosis.
Other Disorders
Magnesium depletion, induced either by dietary restriction or by abnormal loss, reduces the intracellular potassium concentration and causes renal potassium wasting.49 Magnesium depletion often coexists with potassium depletion as a result of drugs (e.g., diuretics and amphotericin B) or disease processes (e.g., hyperaldosteronism and diarrhea) that cause loss of both ions, making it difficult to assess whether the hypokalemia is caused by the hypomagnesemia or is an independent effect. Regardless of the cause, the ability to correct potassium deficiency is impaired when magnesium deficiency is present.
Severe and often refractory hypokalemia due to renal potassium wasting occurs in patients with acute myelogenous, monomyeloblastic, or lymphoblastic leukemia.41
In uncontrolled diabetes mellitus, renal glucose loss causes osmotic diuresis, increasing sodium delivery to the distal nephron and promoting potassium excretion. With prolonged glycosuria, there is considerable depletion of body stores of potassium, but hypokalemia usually is mild or absent because both hypertonicity and insulin deficiency impede the entry of potassium into cells. The underlying potassium deficiency is rapidly unmasked when insulin is given, and severe hypo-kalemia can develop, particularly in patients with diabetic ketoacidosis, unless aggressive replacement of potassium stores is undertaken at the same time.
Severe hypokalemia (serum potassium, < 3.0 mEq/L) can occur, although rarely, in association with hyperthyroidism, which results in a clinical syndrome characterized by the sudden onset of severe muscle weakness and paralysis.41 The symptoms respond rapidly to the administration of potassium.
Familial hypokalemic periodic paralysis is a rare autosomal dominant disease that has been associated with mutations of the gene encoding the dihydropyridine receptor, a voltage-gated calcium channel.50 The disorder is characterized by sudden attacks of muscle paralysis associated with a decrease in serum potassium to low concentrations, often less than 2.5 mmol/L. Attacks can be provoked by high intake of carbohydrates or sodium or by exertion and usually subside spontaneously in fewer than 24 hours. Although the hypokalemia is caused by a shift of potassium into cells, the administration of potassium can be life-saving and should be given to treat acute attacks. Various approaches have been used to prevent recurrences with varying degrees of success, including the administration of spironolactone, triamterene, and acetazolamide.51
Principles of Potassium Replacement
Potassium replacement is the cornerstone of therapy for hypokalemia. Unfortunately, supplemental potassium administration is also the most common cause of severe hyperkalemia in patients who are hospitalized, and this risk must be kept in mind when one is initiating treatment. The risk is greatest with the administration of intravenous potassium, which should be avoided if possible.
The treatment of hypokalemia includes minimizing further potassium loss and giving potassium replacement. IV administration of potassium is indicated when arrhythmias are present or hypokalemia is severe (K+ < 2.5 mEq/L).
Acute potassium administration may be empirical in emergent conditions. When indicated, maximum IV K+ replacement should be 0.31.0 mEq/kg/hr (max 40 mEq/hr) with continuous ECG monitoring during infusion. Central or peripheral IV sites may be used. A more concentrated solution of potassium may be infused if a central line is used, but the catheter tip should not extend into the right atrium.
If cardiac arrest from hypokalemia is imminent (i.e., malignant ventricular arrhythmias), more rapid replacement of potassium is required.36 In the patient’s chart, document that rapid infusion is intentional in response to life-threatening hypokalemia. Once the patient is stabilized, reduce the infusion to continue potassium replacement more gradually.
Calcium
Calcium is an essential ion for function of all cells in the body. It is the primary regulator of motion and regulates excitation-contraction coupling, neurotransmission, hormonal secretion, mitosis, ciliary motion, phagocytosis, and many other processes. These are all vital cell functions which are essential for cellular health.52
In the serum, it consists of three fractions: 1) ionized or free calcium, accounting for 50% of total calcium; 2) protein-bound calcium which is not filterable by the kidney, accounting for approximately 40%; and 3) calcium complexed to anions such as bicarbonate, citrate, sulfate, phosphate, and lactate, accounting for the remaining 10%. Ionized calcium is the physiologically active portion. Aside from serum protein concentration (principally albumin), pH also influences protein binding of calcium and, thus, the ionized calcium level. When clinically available, the ionized calcium level should be followed.
Abnormalities in serum calcium concentration has profound effects on neurological, gastrointestinal, and renal function. Maintenance of the normal serum calcium is a result of tightly regulated ion transport by the kidney, intestinal tract, and bone, mediated by calcemic hormones, especially parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D[1,25(OH)2 D].52,53 Abnormalities in calcium transport that result in uncompensated influx into, or efflux from, the extracellular fluid, will result in hypercalcemia or hypocalcemia, respectively.
Hypocalcemia
Hypocalcemia generally is defined as an ionized calcium level less than 4 mg/dL (1 mmol/L). Hypocalcemia occurs when there is a net efflux of calcium from the ECF; calcium is lost from the ECF, often through renal mechanisms, in greater quantities than can be replaced by intestinal transport or bone. Falsely low levels of calcium due to hypoalbuminemia should be excluded by measuring ionized calcium.
In many cases, hypocalcemia is a result of an inability to mobilize calcium from the skeletal system. This is secondary to decreased secretion of PTH (hypoparathyroidism, hypomagnesemia), impaired synthesis of 1,25(OH)2 D (renal failure, vitamin D-dependent rickets), or inadequate responsiveness of target organs to PTH (pseudohypoparathyroidism, vitamin D deficiency, osteomalacia, renal failure, hypomagnesemia).53
Hypocalcemia, both total and ionized, is common in critically ill children.54-57 The cause of hypocalcemia is uncertain and likely to be multifactorial. In some critically ill children with ionized hypocalcemia, the PTH concentrations are raised whereas in others it is not.54 This suggests the etiology of hypocalcemia is twofold: in some children, a failure to increase PTH concentrations is responsible, and in others, a failure of PTH to elevate calcium levels is the culprit. Low magnesium concentrations may prevent PTH secretion and interfere with its peripheral action leading to hypocalcemia.52 Hypocalcemia may also occur during administration of citrate-buffered blood products or plasma expanders due to free calcium binding with citrate and protein, respectively. Trauma, especially with crush injuries to major muscle groups causing rhabdomyolysis, releases cellular phosphorous which complexes with calcium and lowers serum calcium. Pancreatitis leads to the release of pancreatic lipase, degradation of retroperitoneal omental fat, and binding of calcium in the peritoneum and this loss of extracellular calcium results in hypocalcemia.
The manifestations of hypocalcemia may be ascribed to increased neuromuscular excitability including numbness and tingling of lips, hands, and toes, carpopedal spasms, irritability, laryngeal stridor, apnea, and generalized tonic clonic seizures. Trousseau’s sign (contractions of the hand muscles induced by decreased blood flow to the extremity), and Chvostek’s sign (spasm of the facial muscles evoked by tapping the facial nerve anterior to the ear) may be present. Decreases in cardiac contractility and systemic vascular resistance result in hypotension. Cardiac manifestations also may consist of a prolonged QT interval which may progress to ventricular fibrillation or heart block.
Severe, symptomatic hypocalcemia should be treated immediately.36 Calcium may be given intravenously as 10% calcium chloride (10-20 mg/kg/dose) or 10% calcium gluconate (50-100 mg/kg/dose) by slow infusion. The slow intravenous administration of calcium supplementation (10-15 minutes) and continuous ECG recording are critical to monitor for bradycardia or ventricular irritability. Intravenous access must be secure, as calcium infiltration can result in phlebitis and tissue necrosis. Except in the setting of life-threatening hypocalcemia, calcium salts are generally best administered into a central vein. Multiple doses must be guided by frequent ionized calcium determinations.
Several general principles apply to the management of a hypocalcemic patient. The magnesium level should be checked and, if low, corrected. In a setting of sepsis or renal failure, metabolic acidosis may accompany hypocalcemia and calcium must be replaced before the acidosis is corrected. Calcium and hydrogen ions compete for protein-binding sites so an increase in pH with alkali therapy will increase the binding sites for calcium, leading to a rapid fall in ionized calcium, potentially resulting in cardiac arrest—unless the calcium is corrected first. Sodium bicarbonate and calcium salts must be infused in separate lines to avoid precipitation of calcium carbonate. Patients on digoxin should be monitored carefully because administration of calcium may potentiate digitalis toxicity and cause death. Intravenous calcium should be given cautiously in the presence of hyperphosphatemia; a total calcium times phosphate product of greater than 70 mg/dL may lead to soft-tissue calcification. Similarly, aggressive replacement of phosphorus may precipitate tetany.
Finally, the administration of calcium to critically ill patients is controversial. Available data suggests that decreased ionized calcium levels are protective during critical illness, especially shock states. Administration of calcium to animals with infection increases ionized calcium levels into the normal range and improves blood pressure; however, it also causes increased mortality.54 A variety of mechanisms by which calcium may be detrimental have been suggested.36 Free intracellular calcium concentrations are elevated during critical illness. Unregulated increases in intracellular calcium levels cause cell dysfunction. There is activation of intracellular digestive enzymes (i.e., proteases, lipases, nucleases), increased membrane permeability, impaired mitrochondrial function and ATP production, lysosomal destabilization, impaired cardiac contractility, catecholamine resistance, free radical production, and activated apoptosis. Exogenous calcium administration aggravates these processes.
Hypercalcemia
In pediatrics, hypercalcemia is an uncommon occurrence. This discussion excludes hypercalcemia in the newborn. Hypercalcemia is defined as a measured total serum calcium concentration of greater than 11.0 mg/dL. Hypercalcemia can result from increased calcium absorption from the gut or increased calcium resorption from bone. Hypercalcemia occasionally results from a massive increase in dietary calcium intake or a reduction in the renal excretion of calcium. In hyperparathyroidism, increased bone resorption of calcium results in hypercalcemia, and decreased renal reabsorption of phosphorus results in hypophosphatemia.52,53 Because PTH stimulates bone turnover, alkaline phosphatase is elevated. PTH is inappropriately elevated for the level of serum calcium and confirms the diagnosis.
The vitamin D toxicity syndromes usually can be suspected from the history; hypercalcemia is the result of increased calcium absorption. If the intoxicating compound is conventional vitamin D, then hypercalcemia may be prolonged because of the storage of this compound in adipose tissue.
Symptoms of hypercalcemia may be attributed to its depressive effects on neuromuscular function.52 These include anorexia, nausea, vomiting, lethargy, muscular weakness, confusion, and stupor. Electrocardiographic changes (shortening of the QT interval) may be seen.
Treatment of hypercalcemia is facilitated by knowing the underlying cause of the derangement. The choice of therapy depends on whether the kidneys are functioning normally. The initial emergency treatment of symptomatic hypercalcemia is designed to enhance calcium excretion by saline infusion at a rate of twice maintenance followed by bolus injections of furosemide, 1-2 mg/kg every 6-8 hours. The subsequent amount and rate of saline to be administered depends on the state of hydration and presence or absence of hypertension or preexisting cardiac disease, but in an otherwise normal patient, saline flow rates of two to three times daily maintenance would be appropriate until the serum calcium returns to normal. Treatment of a hypercalcemic crisis depends on the underlying cause, the level of serum calcium, and the severity of signs and symptoms. It always requires hospitalization in an intensive care setting. In acute oliguric renal failure, peritoneal or hemodialysis against a low calcium dialysate is usually effective.
For hypercalcemia that is poorly responsive to conventional treatment, treatment with biphosphonates should be considered.58 Most pediatric experience is with pamidronate: a single dose of 0.5-1 mg/kg IV should normalize the serum calcium in 2-5 days. Hypersensitivity to the drug or its components is the only contraindication to IV administration. It should be used cautiously in renal insufficiency and adequate hydration and urinary output should be ensured during treatment.
Magnesium
Magnesium is the fourth most abundant cation in the body and the second most abundant intracellular electrolyte. Ninety-nine percent is found in the intracellular compartment, and the remaining 1% is in ECF. This important ion is a dependent cofactor in the function of more than 300 enzyme systems.59,60 It is an obligatory cofactor in reactions involving adenosine triphosphate, including maintenance of normal intracellular potassium by the sodium-potassium adenosine triphosphatase pump. It is essential to oxidative phosphorylation, protein synthesis, amino acid activation, and glucose use.59,60 Serum magnesium levels are regulated by the kidneys. The normal serum magnesium ranges between 1.7-2.2 mg/dL (0.75-0.95 mmol/L; 1.5-1.9 mEq/L).
The plasma magnesium concentration is composed of bound and unbound fractions in a manner that is similar to that of calcium; approximately 50% of the circulating magnesium is free (i.e., ionized). In critically ill patients the total magnesium concentration may poorly reflect the physiological (ionized) concentration, the latter can be measured with ion-selective electrodes.61
Particularly in pharmacological concentrations, magnesium can inhibit calcium channels, which represents some of the potentially therapeutic effects of magnesium. Through inhibition of calcium channels and the subsequent reduction of intracellular calcium concentration, magnesium causes smooth muscle relaxation, which has been used in the treatment of acute severe asthma.62 In addition, the effects of magnesium on calcium channels, and perhaps other membrane effects, have been useful in the treatment of torsades de pointes VT.36,60
Hypomagnesemia
In critically ill patients, estimates of magnesium deficiency range from 20%-65% and have been associated with increased mortality rates.60,63 Because magnesium deficiency is common and may lead to life-threatening complications, total serum magnesium concentration has been viewed as the "fifth electrolyte" needed in every patient, in addition to sodium, potassium, chloride, and bicarbonate.63 The usual cause of hypomagnesemia is loss of magnesium from the gastrointestinal tract or the kidney.
Most of the symptoms of moderate to severe hypomagnesemia are non-specific and symptomatic magnesium depletion usually is associated with additional ion abnormalities such as hypocalcemia, hypokalemia, and metabolic alkalosis.64
Hypocalcemia is typical in severe hypomagnesemia, and its degree seems to be related to the severity of the magnesium depletion, usually appearing at a serum below 1.0 mEq/L (0.5 mmol/L.)60 Patients may present with evidence of neuromuscular hyperexcitability, with positive Chvostek and Trousseau signs or spontaneous carpopedal spasm. Most hypocalcemic/hypomagnesemic patients have a low or normal PTH concentration, suggesting impaired synthesis or secretion of PTH. Magnesium supplementation leads to a rapid rise in plasma PTH. Many observations are compatible with a primary role for PTH resistance, where severe hypomagnesemia may alter signal transduction from the PTH receptor to catalytic adenylate cyclase.65
Hypokalemia also is a frequent feature of magnesium deficiency (40-60% of cases). In magnesium deficiency potassium secretion in the loop of Henle and the cortical collecting tubule increases. The hypokalemia here does not respond to potassium replacement, and the magnesium deficit itself has to be corrected.66
Magnesium is vital to carbohydrate metabolism and the generation of both anaerobic and aerobic energy, and it influences glucose catabolism and insulin sensitivity.
Magnesium depletion may produce acute electrocardiographic changes such as widening of the QRS complex and the appearance of peak T waves. In severe depletion the PR interval is prolonged, with progressive widening of the QRS, T wave inversion, and the appearance of U waves. Hypomagnesemia has been implicated in severe ventricular arrhythmias, especially during myocardial ischemia and cardiopulmonary bypass procedures. There also is an association between hypomagnesemia and sensitivity to cardiac glycosides.
The use of intravenous magnesium should be considered for refractory arrhythmias, even in the presence of normal serum magnesium levels, particularly if the patient is on digoxin and diuretics.67 According to numerous case reports, magnesium sulfate has proven effective in treating a wide range of cardiac arrhythmias, especially ventricular arrhythmias, after conventional therapies have failed.68 (See Table 2.)
| Table 2. Cardiac Arrhythmias Terminated by Administration of IV Magnesium Sulfate |
| • Torsade de pointes • Refractory ventricular fibrillation • Refractory ventricular tachycardia • Refractory PVCs • Ventricular arrhythmia associated with digitalis overdose • Multifocal atrial tachycardia |
|
|
The choice of route of magnesium repletion varies with the severity of the clinical findings. An acute infusion of magnesium could decrease magnesium reabsorption in the loop of Henle, most of the infused magnesium ending up in the urine. For this reason, oral replacement is preferred, especially in symptom-free patients.
Symptomatic magnesium deficiency should be treated parenterally. Magnesium sulfate can be supplemented at an IV dose of 25-50 mg/kg/dose (maximum single dose 2 grams) infused over 3-4 hours. Electrocardiographic monitoring is required during intravenous administration of magnesium because hypotension, arrhythmias and skeletal muscle weakness with respiratory failure have been reported (see section on hypermagnesium). Calcium should be readily available as an antidote. Concomitant hypocalcemia and hypokalemia should be corrected separately.
Hypermagnesemia
Hypermagnesemia is uncommon in pediatrics but, as in adults, usually is related to accidental overdose.59,69,70 This is particularly true in patients with impaired renal function. Magnesium-containing over-the-counter medications, particularly antacids and cathartics, may result in hypermagnesium in infants and small children.69 Measurement of serum magnesium concentration is appropriate in the child with unexplained lethargy, hypotonia, respiratory depression, or hypotension.
Manifestations of hypermagnesemia are based on this ion’s affect on the central nervous system (CNS), cardiovascular system, and neuromuscular junction.60 CNS manifestations include drowsiness, confusion, lethargy, and coma. Effects on the cardiovascular system include hypotension and dysrhythmias. These effects are caused by the calcium channel-blocking properties of magnesium, which decreases entry of calcium into cells and enhances egress of calcium from cells. At the neuromuscular junction, magnesium sulfate decreases the amount of acetylcholine liberated, diminishes the sensitivity of the endplate to acetylcholine, and depresses the excitability of the muscle membrane. This results in skeletal muscle weakness and respiratory distress. Clinical signs and symptoms correlate with plasma concentrations.60,69 EKG changes (prolongation of the PR interval, increased duration of the QRS complex, and increased height of the T waves) are noted with concentrations of 6-12 mg/dL (5-10 mEq/L), deep tendon reflexes are lost when serum concentrations reach 12 mg/dL (10 mEq/L), respiratory paralysis and sinoatrial and atrioventricular block occurs at 18 mg/dL (15 mEq/L), and cardiac arrest occurs at 30 mg/dL (25 mEq/L).
Management of hypermagnesemia is dictated by the cardiovascular, CNS, and neuromuscular changes that occur. Treatment consists of stopping the source of magnesium, respiratory and cardiovascular support, calcium, diuresis, and dialysis. Indications for dialysis include: kidney failure, increasing magnesium levels despite diuresis, dysrhythmias, and persistent hemodynamic instability.
References
1. Rothrock SG, Green SM, McArthur CL, et al. Detection of electrolyte abnormalities in children presenting to the emergency department: A multicenter, prospective analysis. Acad Emerg Med 1997; 4:1025-1031.
2. Jospe N, Forbes G. Fluids and electrolytes clinical aspects. Pediatrics in Review 1996;17:395-403.
3. Kumar S, Berl T. Sodium. Lancet 1998;352:220-228.
4. Lee C, Guo H, Chen J. Hyponatremia in the emergency department. Am J Emerg Med 2000;18:264-268.
5. Arieff AI, Ayus JC, Fraser CL. Hyponatremia and death or permanent brain damage in healthy children. BMJ 1992;304:1218-1222.
6. Arieff AI, Ayus JC. Pathogenesis of hyponatremic encephalopathy: Current concepts. Chest 1993;103:607-610.
7. Jackson J, Bolte RG. Risks of intravenous administration of hypotonic fluids for pediatric patients in ED and prehospital settings: Let’s remove the handle from the pump. Am J Emerg Med 2000;18:269-270.
8. Bhalla P, Eaton FE, Coulter JGS, et al. Hyponatremic seizures and excessive intake of hypotonic fluids in young children. BMJ 1999;319:1554-1557.
9. Arieff AI, Kronlund BA. Fatal child abuse by forced water intoxication. Pediatrics 1999;103:1292-1295.
10. Farrar HC, Chande VT, Fitzpatrick DF, et al. Hyponatremia as the cause of seizures in infants: A retrospective analysis of incidence, severity, and clinical predictors. Ann Emerg Med 1995;26:42-48.
11. Gold I, Koenigsberg M. Infantile seizures caused by voluntary water intoxication. Am J Emerg Med 1986;4:21-23.
12. Corneli HM, Gormley CJ, Baker RC. Hyponatremia and seizures presenting in the first two years of life. Pediatr Emerg Care 1985;1:190-193.
13. Krugman SD, Zorc JJ, Walker AR. Hyponatremic seizures in infancy: Association with retinal hemorrhages and physical child abuse? Pediatr Emerg Care 2000;16:432-434.
14. Keating JP, Schears GJ, Dodge PR. Oral water intoxication in infants. Am J Dis Child 1991;145:985-990.
15. Avner ED. Clinical disorders of water metabolism: Hyponatremia and hypernatremia. Pediatr Ann 1995;24:23-30.
16. Oster JR, Singer I. Hyponatremia, hypoosmolality and hypotonicity. Arch Intern Med 1999;159:333-336.
17. Bohn D. Problems associated with intravenous fluid administration in children: Do we have the right solutions? Curr Opin Pediatr 2000;12:217-221.
18. Oh MS, Carroll HJ. Cerebral salt-wasting syndrome: We need better proof of its existence. Nephron 1999;82:110-114.
19. Ganong CA, Kappy MS. Cerebral salt wasting in children: The need for recognition and treatment. AJDC 1993;147:167-169.
20. Harrigan MR. Cerebral salt wasting syndrome: A review. Neurosurgery 1996;38:152-160.
21. Oruckaptan HH, Ozisik P, Akalan N. Prolonged cerebral salt wasting syndrome associated with intraventricular dissemination of brain tumors. Pediatr Neurosurg 2000;33:16-20.
22. Kappy MS, Ganong CA. Cerebral salt wasting in children: The role of atrial natriuretic hormone. Advances in Pediatrics 1996;43:271-307.
23. Levine LS. Congenital adrenal hyperplasia. Pediatrics in Review 2000;21:159-170.
24. Sarnaik AP, Meert K, Hackbarth R, et al. Management of hyponatremic seizures in children with hypertonic saline: A safe and effective strategy. Crit Care Med 1991;19:758-762.
25. Hoover B, Gebara BM. Hypertonic 3% saline infusion by nasogastric tube for the treatment of severe symptomatic hyponatremia. Clin Pediatr 1999;38:55-57.
26. Porzio P, Halberthal M, Bohn D, et al. Treatment of acute hyponatremia: Ensuring the excretion of a predictable amount of electrolyte-free water. Crit Care Med 2000;28:1905-1910.
27. Berl T. Mannitol: A therapeutic alternative in the treatment of acute hyponatremia? Crit Care Med 2000;28:2152-2153.
28. Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med 2000;342:1493-1499.
29. McManus ML, Churchwell KB, Strange K. Regulation of cell volume in health and disease. N Engl J Med 1995;333:1260-1266.
30. Baugh JR, Krug EF, Weir MR. Punishment by salt poisoning. Southern Medical Journal 1983;76:540-541.
31. Saborio P, Tipton GA, Chan JCM. Diabetes insipidus. Pediatrics in Review 2000;21:122-129.
32. Ng PC, Chan HB, Fok TF, et al. Early onset of hypernatremic dehydration and fever in exclusively breast-fed infants. J Paediatr Child Health 1999;35:585-587.
33. Halperin ML, Kamel KS. Potassium. Lancet 1998;352:135-140.
34. Roberts KB. Hyperkalemia. Pediatrics in Review 1996;17:106.
35. Dittrich K, Walls R. Hyperkalemia: ECG manifestations and clinical considerations. J Emerg Med 1986;4:449-455.
36. American Heart Association. Advanced challenges in resuscitation. Section 1: Life-threatening electrolyte abnormalities. Circulation 2000;102(Suppl I):I-217-I-222.
37. Ahee P, Crowe AV. The management of hyperkalemia in the emergency department. J Accid Emerg Med 2000;17:188-191.
38. Kim HJ. Combined effect of bicarbonate and insulin with glucose in acute therapy of hypokalemia in end-stage renal disease patients. Nephron 1996;72:476-482.
39. McDonald RA. Disorders of potassium balance. Pediatr Ann 1995;24:31-36.
40. McClure R, Prasad V, Brocklebank J. Treatment of hyperkalemia using intravenous and nebulized salbutamol. Arch Dis Child 1994;70:126-128.
41. Gennari FJ. Hypokalemia. N Engl J Med 1998;339:451-458.
42. Clausen T, Everts ME. Regulation of the Na, K-pump in skeletal muscle. Kidney Int 1989;35:1-13.
43. Blumberg A, Wiedmann P, Ferrari P. Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure. Kidney Int 1992;41:369-374.
44. Field MJ, Giebisch GJ. Hormonal control of renal potassium excretion. Kidney Int 1985;27:379-387.
45. Bello O, Sehabiague G. Hypokalemic paralysis. Pediatr Emerg Care 1998;14:409-410.
46. Naparstek Y, Gutman A. Spurious hypokalemia in myeloproliferative disorders. Am J Med Sci 1984;288:175-177.
47. Leikin JB, Linowiecki KA, Soglin DF, et al. Hypokalemia after pediatric albuterol overdose. A case series. Am J Emerg Med 1994;12:64-66.
48. Schurman SJ, Shoemaker LR. Bartter and Gitelman Syndromes. Advances in Pediatrics 2000;47:223-248.
49. Kobrin SM, Goldfarb S. Magnesium deficiency. Semin Nephrol 1990;10:525-535.
50. Sillen A, Sorenson T, Kantola I, et al. Identification of mutations in the CACNLIA3 gene in 13 families of Scandinavian origin having hypokalemic paralysis. Am J Med Genet 1997;69:102-106.
51. Stedwel RE, Allen KM, Binder LS. Hypokalemic paralyses: A review of the etiologies, pathophysiology, presentation, and therapy. Am J Emerg Med 1992;10:143-148.
52. Bushinsky DA, Monk RD. Calcium. Lancet 1998;352:306-311.
53. Marx JJ. Hyperparathyroid and hypoparathyroid disorders. N Engl J Med 2000;343:1863-1875.
54. Baines PB, Thomson APJ, Fraser WD, et al. Hypocalcemia in severe meningococcal infections. Arch Dis Child 2000;83:510-513.
55. Zaloga GP. Hypocalcemia in critically ill patients. Crit Care Med 1992;20:251-262.
56. Gauthier B, Trachtman H, DiCarmine F, et al. Hypocalcemia and hypercalcitonemia in critically ill children. Crit Care Med 1990;18:1215-1219.
57. Cardenas-Rivero N, Chernow B, Stoiko MA, et al. Hypocalcemia in critically ill children. J Pediatr 1989;114:946-951.
58. Srivastava T and Alan US. Biphophonate: From grandparents to grandchildren. Clin Pediatr 1999;38:687-702.
59. Harker HE, Majcher TA. Hypermagnesemia in a pediatric patient. Anesth Analg 2000;91:1160-1162.
60. Weisinger JR, Bellorin-Font E. Magnesium and phosphorus. Lancet 1998;352:391-396.
61. Fiser RT, Torres A, Butch AW, et al. Ionized magnesium concentrations in critically ill children. Crit Care Med 1998;26:2048-2052.
62. Ciarallo L, Brousseau D, Reinert S. Higher-dose intravenous magnesium therapy for children with moderate to severe acute asthma. Arch Pediatr Adolesc Med 2000;154:979-983.
63. Hebert P, Mehta N, Wang J, et al. Functional magnesium deficiency in critically ill patients identified using a magnesium-loading test. Crit Care Med 1997;25:749-755.
64. Rude RJ. Magnesium metabolism and deficiency. Endocrinol Metab Clin N Am 1993;22:377-395.
65. Abbott LG, Rude RK. Clinical manifestations of magnesium deficiency. Mineral Electrolyte Metab 1993;19:314-322.
66. Ryan MP. Interrelationship of magnesium and potassium homeostasis. Mineral Electrolyte Metab 1993;19:290-295.
67. Keller KB, Lemberg L. The importance of magnesium in cardiovascular disease. Am J Crit Care 1993;2:348-350.
68. Dickinson ET. Magnesium comes of age. Journal of Emergency Medical Services (JEMS) 1992;17:60-61.
69. Sullivan JE. Hypermagnesemia with lethargy and hypotonia due to administration of magnesium hydroxide to a 4 week old infant. Arch Pediatr Adolesc Med 2000;154:1272-1274.
70. Vissers RJ, Purssell R. Iatrogenic magnesium overdose: Two case reports. J Emerg Med 1996;14:187-191.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.