Idiopathic Inflammatory Myopathy: A Review and Update
Idiopathic Inflammatory Myopathy: A Review and Update
Authors:
Kavoos Nazeri, MD, Fellow of Rheumatology, Loma Linda University, Loma Linda, CA
Keith Colburn, MD, Professor of Medicine, Chief of Rheumatology, Loma Linda University and Loma Linda VA Medical Center, Loma Linda, CA
Peer Reviewer:
Sharon Merryman, DO, Dayton Center for Neurological Disorders, Dayton, OH
The idiopathic inflammatory myopathies are a heterogeneous group of autoimmune syndromes characterized by subacute or chronic muscle weakness and skeletal muscle inflammation. Of the idiopathic inflammatory myopathies, the best recognized subsets are polymyositis, dermatomyositis, inclusion body myositis (IBM), and the newly described autoimmune necrotizing myopathy. The less common inflammatory myositis diagnoses include granulomatous myositis (sarcoidosis), eosinophilic myositis, infectious myositis, and the myositis associated with overlap syndromes.1,2
This article will review the immmunopathogenesis, clinical features, diagnosis, and management of the most common idiopathic inflammatory myopathies including polymyositis, dermatomyositis, IBM, and autoimmune necrotizing myopathy.
Epidemiology
The idiopathic inflammatory myopathies are rare disorders. The overall annual incidence is approximately 1 in 100,000. They affect women more than men, except for inclusion body myositis where the male to female ratio may be as high as 3:1. Interestingly, the age-adjusted prevalence of IBM in people older than 50 years of age is about 3.5/100,000, which makes it the most common autoimmune myopathy in this age group, representing 16-28% of all myopathies.3
The incidence of the inflammatory myopathies varies with age and ethnicity. Some studies report a higher incidence of polymyositis in African Americans. The onset of polymyositis usually occurs in the late teens or in older patients (mean age at onset in the 50s). Dermatomyositis shows two peaks: 5-15 years and 45-65 years. IBM usually is seen in individuals older than 50 and is rare in younger adults.4
A significant portion (11-44%) of inflammatory myopathies can occur in association with other autoimmune disorders, such as scleroderma, systemic lupus erythematosus (SLE), mixed connective tissue disease, rheumatoid arthritis, Sjogren's syndrome, polyarteritis nodosa, and sarcoidosis.
Several studies describe an association of malignancies with inflammatory myopathies. The frequency of myopathies associated with malignancies varies widely (4-42%) in different studies but in general, the incidence of malignancy is higher in dermatomyositis patients compared with polymyositis and IBM patients.5
Classification
Several classifications have been proposed for idiopathic inflammatory myopathies. The original Bohan and Peter criteria were formulated in 1975 based on the following features:
- Symmetrical proximal muscle weakness
- Typical rash of dermatomyositis
- Myopathic changes on electromyography
- Characteristic muscle biopsy
- Elevated muscle enzymes
These criteria were formulated before discovery of muscle-specific autoantibodies. This classification does not include IBM, which was not recognized until 1980.6 A second classification was proposed by international workshop of myositis experts in 2004 based on clinical findings, histopathology, and laboratory findings. The new classification excludes myositis associated with other connective tissue disease but includes the following categories:7
- IBM
- Probable and definite polymyositis
- Probable and definite dermatomyositis
- Amyopathic dermatomyositis, also known as dermatomyositis sine myositis
- Possible amyopathic dermatomyositis
- Non-specific myositis
- Immune-mediated necrotizing myopathy
See Table 1 for clinical and laboratory findings related to idopathic inflammatory myopathies.

Pathogenesis
Although the etiology and pathogenesis of idiopathic inflammatory myopathies remains unclear, a number of lines of investigations suggest possible ways in which selected environmental exposures in genetically susceptible individuals may lead to chronic immune activation and an immunologic attack on muscle and other involved tissues.
Common immune activation processes in muscle include up-regulation of MHC class I expression and IL-1 alpha and beta, leading to autoantibody production prior to the onset of clinical disease. Then myocyte-directed cytotoxic T-cell mechanisms predominate in polymyositis and IBM, while complement-mediated endothelial damage leading to CD4, B-cell, and dendritic cell infiltration predominates in muscle tissue in dermatomyositis. Other possible disease mechanisms may include hypoxia, activation of endoplasmic reticulum stress response, and cleavage of auto-antigens resulting in cytokine and chemokine release. Later processes include muscle regeneration, angiogenesis, repair, and, in some cases, fibrotic changes.8,9
Clinical Features
Polymyositis and Dermatomyositis
Polymyositis and dermatomyositis are multisystem disorders with a wide variety of clinical manifestations.4 The most prominent findings in different organs of the body are described below.
Muscle. Muscle weakness is the most common presenting feature of dermatomyositis and polymyositis. The onset is usually insidious with gradual worsening over a period of several months. The distribution of the weakness is typically symmetrical and in the proximal muscles. Distal muscle weakness, if present, tends to be mild. Rarely, some patients may present with focal muscle weakness that later progresses to a more generalized form of myositis over time.
Myalgia and muscle tenderness occur in 25-50% of patients. These symptoms tend to be milder in polymyositis and dermatomyositis compared to the pain of polymyalgia rheumatica, fibromyalgia, or viral myositis. Muscle atrophy usually is not seen in early stages but may occur in severe, longstanding disease.4
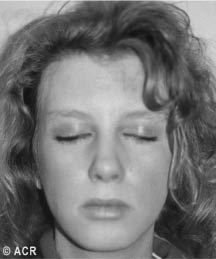
Figure 1: Characteristic Features of DM Skin Changes
Printed with permission from the American College of Rheumatology slide collection
A. Heliotrope rash
B. Gottron's papules
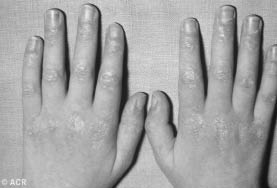
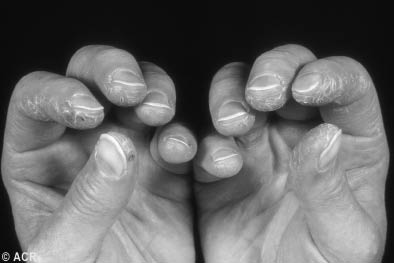
Figure 2: Mechanics Hand in Patient with Dermatomyositis
Printed with permission from the American College of Rheumatology slide collection
Skin. Several distinct rashes have been described in dermatomyositis but not in polymyositis. (See Figures 1 and 2.)
- Gottron's sign refers to an erythematous, scaly eruption that occurs in a symmetrical fashion over the extensor surfaces of the metacarpophalangeal and interphalangeal joints. Similar lesions also can be seen on the extensor aspects in elbows and knees, mimicking psoriasis.
- Heliotrop rash is a term for a violaceous eruption in the upper eyelids, often accompanied by eyelid swelling.
- Shawl sign or V sign is a diffuse photosensitive, erythematous rash over the upper chest and neck in a V-shaped distribution.
- Capillary nail bed changes similar to those seen in scleroderma and the overlap syndromes also are observed in dermatomyositis showing alternating areas of dilation of capillary loops and capillary dropout.
- Mechanic's hand is described in both dermatomyositis and polymyositis. It is evident by roughening and cracking of the skin of the tips and lateral sides of fingers.
- Psoriasiform-like changes of the scalp occur frequently in patients with dermatomyositis.
- Flagellate erythema comprised of linear, violacious streaks occurs on the trunk. Recurrent scratching of skin is believed to play a role in the etiology of these lesions.
- Calcinosis, including calcinosis cutis, is the deposition of calcium in the skin, and calcinosis universalis is the deposition of calcium in sheets in the muscles and tendons. These occur commonly in juvenile dermatomyositis, but have also been reported in adult dermatomyositis.
- Other rare skin manifestations of dermatomyositis include icthyocytosis, panniculitis, lichen planus, vesicle and bullae formation, follicular hyperkeratosis, and papular mucinosis.
Lungs. Interstitial lung disease (ILD) is an important complication of polymyositis and dermatomyositis, and is a leading cause of death in these two diseases. At least 10% of patients with dermatomyositis and polymyositis will develop interstitial lung involvement. Eighty percent of patients with anti-Jo-1 antibodies in their serum will have ILD. The occurrence of ILD also often is associated with antisynthetase antibodies and the antisynthetase syndrome. Respiratory muscle involvement can happen in advanced stages of inflammatory myopathies resulting in patients needing mechanical ventilation. Involvement of respiratory muscles in these patients is associated with a poor prognosis.
Esophagus. The upper one-third of the esophagus is largely composed of striated muscle and, thus, is vulnerable to the inflammatory activity of polymyositis or dermatomyositis. Weakness of the muscles of the oropharynx and the esophagus may lead to dysphagia, nasal regurgitation, and/or aspiration. Esophageal involvement is more common in the elderly and increases the chance of aspiration pneumonia.
Cardiac. Myocarditis is well described in polymyositis and dermatomyositis. However, myocardial involvement severe enough to cause heart failure is unusual. Patients with cardiac involvement may have increased serum levels of CK-MB and troponin in addition to increased routine CK levels.
Antisynthetase Syndrome. Up to 30% of patients with polymyositis and dermatomyositis may develop a constellation of clinical symptoms termed antisynthetase syndrome. These findings include relative acute disease onset, constitutional symptoms (fever, weight loss, generalized body pain), Raynaud's phenomenon, mechanic's hands, arthritis, and interstitial lung disease.
Most of these patients have antisynthetase antibodies that are highly specific for autoimmune myositis. Some patients with antisynthetase syndrome have relatively little myositis but other more prominent features of this disease spectrum, such as interstitial lung disease.10
Other Features. Patients with polymyositis or dermatomyositis may present with a variety of other manifestations, including fever, weight loss, Raynaud's phenomenon, and non-erosive inflammatory polyarthritis.
Inclusion Body Myositis
IBM is the most common idiopathic inflammatory myopathy in patients age 50 years or older, and it accounts for approximately 30% of all inflammatory myopathies. It is also the most common myopathy misdiagnosed as polymyositis in cases that appear to be refractory polymyositis. Distinction between two conditions is critical because the patient's prognosis differs significantly. In contrast to polymyositis, IBM generally has a more insidious onset and more prominent distal muscle weakness. Furthermore, in many patients with IBM, the muscle involvement is asymmetric, particularly in the beginning. Up to 40% of patients with IBM may have dysphagia at the time of diagnosis. On average, serum muscle enzyme levels are lower in IBM compare to polymyositis, although substantial elevations may occur. The presence of typical inclusion bodies on muscle biopsy is diagnostic for this disorder, but a single biopsy misses the diagnosis in 20-30% of cases. Magnetic resonance imaging may help distinguish polymyositis from IBM. Whereas MRI changes suggestive of inflammation are noted along the fascia planes in polymyositis, such changes are observed throughout the muscle in IBM.11,12
Immune-mediated Necrotizing Myopathy
Immune-mediated (autoimmune) necrotizing myopathy is a unique autoimmune myopathy with distinct pathologic features. It is an increasingly recognized myopathy that pathologically has little or no immune infiltrate on histopathology. Autoimmune necrotizing myopathy presents as a subacute or insidious progressive proximal muscle weakness without a rash. Weakness generally develops more rapidly in comparison to polymyositis and in 30% of cases it is markedly severe. There may be associated myalgia and in some cases dysphagia. In a Dutch study, autoimmune necrotizing myopathy represented 19% of all inflammatory myopathies. It was more common in patients over the age of 30 years old and had a female to male ratio of 3:1.13
Autoimmune necrotizing myopathy commonly is associated with other connective tissue diseases including scleroderma and mixed connective tissue disorder (MCTD). This type of myopathy can be triggered by statin therapy. In predisposed individuals, statin-associated toxic myopathy evolves into an autoimmune myopathy that progresses beyond 3-6 months after statin discontinuation. In one study, the mean age of affected patients was 65.5 years. The onset of autoimmune necrotizing myopathy may be delayed up to 10 years following the initiation of statin therapy.14
Paraneoplastic necrotizing myopathy also has been described as a rare, rapidly progressive, and severe variant of necrotizing myopathies associated with a malignancy that affects adults older than age 40 years.13
Overlap Syndrome
Dermatomyositis and polymyositis may overlap with features of other connective tissue diseases, particularly scleroderma, SLE, and Sjogren's syndrome. These conditions often are referred to as either MCTD if associated with the presence of anti-RNP antibodies or undifferentiated connective tissue disease (UCTD). The myopathy associated with other connective tissue diseases varies from clinically insignificant with minimal elevation of muscle enzymes of biopsy changes to a more severe form of myopathy like polymyositis or dermatomyositis.15
Diagnosis
The diagnosis of idiopathic myopathy is suggested by the patient's history and clinical findings as described above. It also is necessary to exclude other causes of muscle weakness and elevated muscle enzymes including but not limited to drug-induced myopathy, infectious myopathy, hypothyroidism, myasthenia gravis, muscular dystrophies, rhabdomyolysis, sarcoidosis, and metabolic myopathies.
The elevation of muscle enzymes, the presence of specific antibodies, and specific EMG and MRI findings can facilitate the diagnosis. The definitive test to establish the diagnosis of idiopathic inflammatory myopathies is an open-muscle biopsy.4
Muscle Enzymes. Creatine kinase (CK) and aldolase are the serum muscle enzymes routinely measured in the evaluation of myopathies. Lactate dehydrogenase (LDH), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) also often are elevated but are not specific for muscle pathology. The serum CK level is increased in the majority of dermatomyositis patients with levels often ranging more than 100 times the upper limit of normal. In fewer than 10% of cases of dermatomyositis, regardless of severity, serum CK levels may be normal. Measurement of the serum aldolase can be helpful since sometimes it may be elevated in absence of an elevation of serum CK. There almost always is an increase in CK levels in polymyositis, like in dermatomyositis, often more than 100 times the normal range. In autoimmune necrotizing myopathy, the serum CK levels may be 10 times the upper limits of normal. The serum levels of CK and aldolase decrease with a good clinical response to treatment but do not correlate with the severity of muscle weakness. Near normal muscle enzyme levels associated with significant weakness in an elderly person suggests IBM or extensive muscle atrophy.16
Autoantibodies. Antinuclear antibodies (ANA) detected by standard immunofluorescence methods are present in 80% of patients with polymyositis and dermatomyositis, but are not specific for either condition. Detection of anti-SS-A (anti-Ro), anti-SS-B (anti-La), anti-Smith, or anti-ribonucleoprotein (anti-RNP) antibodies strongly suggests the diagnosis of myositis associated with overlap syndromes such as MCTD or UCTD.17
Myositis-specific Autoantibodies. Several categories of autoantibodies directed against cytoplasmic RNA synthetase, ribonucleoproteins, and certain nuclear antigens are called myositis-specific autoantibodies. These antibodies are more specific than a serum ANA and occur in approximately 30% of patients with polymyositis and dermatomyositis. It appears increasingly that myositis-specific autoantibodies play a specific role in the pathophysiology of the idiopathic inflammatory myopathies. There are three major categories of these antibodies.18,19 (See Table 2.)

- Anti-Jo-1 antibodies are the most common of the anti-synthetase antibodies directed against anti-histidyl-tRNA synthetase. Anti-Jo-1 antibodies are strongly associated with interstitial lung disease, Raynaud's phenomenon, arthritis, and mechanic's hands.
- Anti-SRP antibodies are directed against signal recognition particles (SRP). These antibodies are mainly detected in polymyositis and are associated with severe, aggressive myopathy.
- Anti-Mi-2 antibodies are against a helicase involved in transcriptional activation. Among patients with dermatomyositis, these antibodies are associated with relatively acute disease onset and significant skin rashes but better overall prognosis.
Electromyography. In addition to laboratory testing, electromyography (EMG) is very important in the evaluation of patients with inflammatory myopathies. The EMG shows evidence of increased membrane irritability as described in the classic triad below:
- Increased insertional activity and spontaneous fibrillation
- Abnormal myopathic low amplitude, short-duration polyphasic motor potentials
- Complex repetitive discharges
A normal EMG is unusual in patients with typical poly or dermatomyositis. EMG abnormalities may support the diagnosis but are not specific for idiopathic inflammatory myopathies.16,20
Magnetic Resonance Imaging. Magnetic resonance imaging (MRI) has emerged as an important technique in clinical evaluation of patients with inflammatory myopathies. MRI can show areas of myositis with inflammatory changes, edema, muscle fibrosis, and calcification. It also helps minimize sampling error for a muscle biopsy, since in many cases there may be localized or a patchy inflammatory involvement of the muscles. MRI also has the additional advantage of permitting serial assessments, which may be useful in the evaluation of a patient's response to therapy (see Figure 3). Another MRI modality is MR spectroscopy, which provides a view of muscle metabolism by comparing the ratio of muscle phosphorus contained in phosphocreatine to the level of inorganic phosphorus. This ratio is decreased in inflammatory myopathies. MR spectroscopy is very sensitive in detecting slight inflammatory changes in muscles. The exact role and utility of this modality requires further study.21
Figure 3: MRI of Thighs in Patient with Localized Myositis of Thigh Muscles
Printed with permission from the American College of Rheumatology slide collection
Muscle Biopsy. For a majority of patients with myositis, the definitive test to establish the diagnosis of inflammatory myopathy is a muscle biopsy. The biopsy should be obtained from a muscle that on physical examination is weak. The usual biopsy sites are quadriceps or deltoid muscle. If possible the biopsy should be done on a muscle that was not penetrated with a needle from an EMG study. A biopsy of the calf muscles is discouraged because it results in frequent histological artifacts and is not a proximal muscle where the disease is most prominent. MRI also can help identify the appropriate biopsy site in cases where the initial biopsy was not adequate to make the diagnosis or the EMG failed to reveal the location of the myopathy. An open biopsy is preferred to closed needle biopsy because a more accurate sample can be obtained and the muscle fibers are better preserved. In one study, open muscle biopsy had overall 83% sensitivity for the diagnosis of polymyositis and dermatomyositis. Repeating the biopsy with MRI can increase the sensitivity of detecting myositis.22
The most characteristic changes seen in the biopsy in polymyositis include degeneration and regeneration of muscle fibers and the presence of CD8+ T lymphocytes invading non-necrotic fibers. In tissue samples from patients with dermatomyositis, immune complex deposition, CD4+ T cells, and B cells invade predominantly the perivascular areas leading to perifascicular atrophy.23
Basophilic-rimmed vacuoles within the muscle fiber sarcoplasm are characteristic of IBM. These typical vacuoles may be absent in the initial biopsy of up to 30% of patients due primarily to processing errors. Eosinophilic inclusions may be found adjacent to basophilic vacuoles. Inclusion bodies are highly specific for IBM but are present only in 50% of patients.11
The most characteristic finding in autoimmune necrotizing myopathy is the presence of scattered necrotic myofibers with a paucity of cellular infiltrates. Additionally, microvascular deposition of complement complexes suggests a humorally mediated microangiopathy. Unlike dermatomyositis, perivascular inflammation is scant.1
Management
The goals of treatment are to arrest muscle inflammation, improve muscle strength, and avoid the development of extramuscular complications. Immunosuppressive therapy is the mainstay of treatment in patients with active idiopathic inflammatory myopathies. In contrast to other inflammatory myopathies, patients with IBM are fairly resistant to standard immunosuppressive therapies. Autoimmune necrotizing myopathy often is more resistant to therapy compared to polymyositis or dermatomyositis, particularly when it is associated with statin therapy or malignancy.
Other factors associated with worse prognosis include delay in diagnosis and treatment, extramuscular involvement, and the presence of some autoantibodies especially the anti-synthetase antibodies. Prompt diagnosis and referring the patients to a specialist experienced in treating these conditions plays an important role in the overall prognosis.1,11
In addition to drug therapy, there are a variety of other important considerations in the treatment of patients with inflammatory myopathies. These include physical therapy and rehabilitation, prophylactic measures to prevent aspiration, avoiding decubitus ulcers and DVTs in bedridden patients, and educating patients with dermatomyositis to avoid UV light and sun exposure.24
Successful management of patients with an idiopathic inflammatory myositis requires appropriate laboratory tests every 6-12 weeks, including a CPK, an aldolase, and a CBC to monitor drug toxicity. We also monitor the ESR, CRP, and urinalysis.
Initial Therapy. Although older studies were unable to demonstrate an improvement in survival with glucocorticoid therapy, there is a general consensus that glucocorticoid therapy improves strength and preserves muscle function in dermatomyositis, polymyositis, and necrotizing myopathy. In one trial, 39% of patients with polymyositis and dermatomyositis had a normalization of muscle enzymes, and 25% regained full muscle strength with steroids alone. In another trial, the full response rate was 64% for necrotizing myopathy and polymyositis.25
There is no standard glucocorticoid therapy regiment to treat these patients. The general rule is to start with high doses of a corticosteroid such as prednisone, 1 mg/kg (up to 80 mg) per day, which may be given in divided doses for the first 1-2 weeks. Pulse methylprednisolone can also be used (1000 mg IV per day for 3 days) in critically ill patients. The initial steroid dose needs to be tapered slowly over a period of 9-12 months. A proposed practical way to manage corticosteroid treatment is to continue the initial dose of 1 mg/kg for 4-6 weeks then taper by 10 mg weekly until a dose of 40 mg daily is reached. Next taper the prednisone by 5 mg per week until reaching a dose of 20 mg per day. After that decrease the prednisone by 2.5 mg per week to the lowest dose one can reach without reactivating the disease while the patient is taking a steroid-sparing medication which will be described below.26
Steroid-sparing Agents. Some clinicians initiate steroid-sparing agents at the same time the treatment with prednisone begins. Others reserve these agents for later in the course of the disease as treatment for steroid-resistant cases. It is our opinion that these agents should be started early to avoid as many of the corticosteroid side effects as possible.
Methotrexate (MTX), an anti-folate agent that inhibits lymphocyte proliferation, is an effective immunomodulating drug for the treatment of most of the inflammatory muscle diseases. Several studies found response rates to MTX in patients with polymyositis and dermatomyositis that previously failed corticosteroids to be between 71-82%.25 Some investigators recommended a starting dose of MTX of 15 mg once weekly given orally or subcutaneously. However, we usually start at 20-25 mg weekly. For doses > 25 mg weekly, it is necessary to give the MTX intramuscularly or subcutaneously for more complete absorption of the drug. The dose may be increased by 5 mg every week. Doses up to 50 mg weekly are used. To protect against toxic effects of MTX, leucovorin 15-25 mg rescue, 12-24 hours after the MTX is taken, is needed for doses higher than 25 mg weekly. The therapeutic effect of MTX usually appears 4-6 weeks after starting the treatment. Most common side effects of MTX include stomatitis, alopecia, liver toxicity, and bone marrow suppression. MTX is not recommended for patients with the complication of interstitial lung disease or possibly those with Jo-1 antibodies due to the potential of worsening pulmonary function.25
Azathioprine (AZA), an antimetabolite that blocks T cell proliferation, is also a very effective steroid-sparing agent. A clinical trial of 16 patients on AZA did not show any additive benefit to corticosteroids in the induction of remission. However, in a 3-year follow-up, the patients on AZA had a better functional outcome and required a much lower dose of corticosteroids to control their disease. The recommended dose for AZA is 2-3 mg/kg/day, and can be given in a single or divided dose. The beneficial effects are usually present by 1-2 months.1,25 It is recommend by some investigators to check a thiopurine methyltransferase (TPMT) level because a deficiency of this enzyme can increase the patient's risk of AZA bone marrow toxicity. A CBC should be done 2-3 weeks after starting AZA to be sure bone marrow toxicity is not present. The most common side effects of AZA include rash, hepatotoxicity, pancreatitis, and bone marrow suppression. A CBC and liver enzymes need to be closely monitored every 1-3 months while on this drug.
It is important for primary care doctors to collaborate with a rheumatologist when caring for patients with inflammatory muscle diseases because of the difficulty of managing these rare and dangerous conditions, and the risks of using long-term corticosteroids and other immunosuppressive therapies. It may be wise to do a PPD skin test and if positive, start isoniazid (INH) or rifampin treatment to avoid activation of latent tuberculosis. A baseline DEXA scan and yearly follow-up scans are important while the patient is taking corticosteroids. It is also recommended that patients start prophylactic calcium (1000-1500 mg/day), vitamin D (400-800 IU daily), and a proton pump inhibitor (PPI) for GI prophylaxis even though the PPI can predispose a patient to osteoporosis. Patients need to be asked regularly about any behavioral changes due to psychiatric side effects of glucocorticoids. The patient's blood pressure and serum glucose levels need to be carefully monitored. It is also recommended that all these patients receive pneumococcal vaccine and yearly flu shots.1
Treatment of Resistant Disease. Multiple options exist for treating patients who do not respond adequately to glucocorticoids in combination with MTX or AZA. Rituximab and IVIG were shown to be highly effective in many patients. Other immunosuppressive drugs such as cyclophosphamide, cyclosporine, and tacrolimus are effective treatment modalities for polymyositis and dermatomyositis, but with a significant side effect profile.
There are several studies using rituximab in patients with idiopathic inflammatory myopathies resistant to conventional treatment. Rituximab is an anti-CD 20 monoclonal antibody that depletes a patient's B cells (except plasma cells) within weeks of infusion. Most studies demonstrated a significant response in a majority of patients within a few weeks of administration.27
IVIG is a pooled gamma-globulin product. It has complex immunomodulatory mechanisms including binding to autoantibodies, inflammatory cytokines suppression, and blocking the FC receptors. IVIG is an effective short-term therapy in the inflammatory myopathies resistant to corticosteroids. A randomized, controlled trial with optional crossover showed that IVIG at a dose of 2 mg/kg administered monthly for 3 months was effective in 9 of 12 patients with polymyositis and dermatomyositis resistant to steroids. More recently IVIG also was shown to be effective in treating autoimmune necrotizing myopathy.28
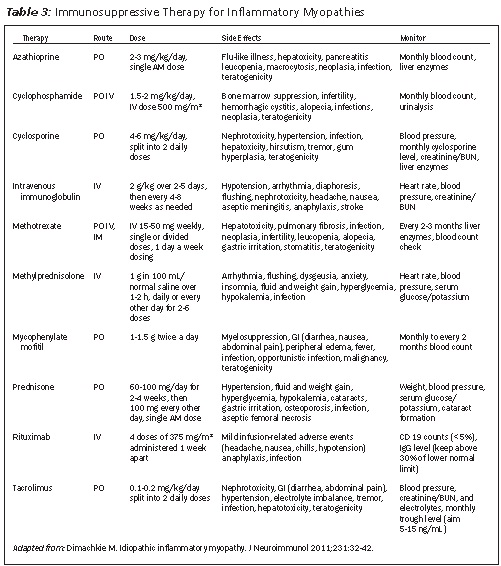
Stem cell transplant clinical trials are being conducted at present and are very promising for patients with life-threatening complications of dermatomyositis and polymyositis. See Table 3 for the most common immunosuppressive therapies, common side effects, and monitoring requirements for inflammatory myopathies.
Assessing Treatment of Inflammatory Myositis. The response to treatment should be assessed every 2-4 weeks after starting the treatment for first 3-6 months then every 2-3 months thereafter. The assessment should include checking muscle strength and assessing the change in muscle enzymes in response to treatment. Improvement in muscle weakness and decline in muscle enzymes usually is achieved within 2 weeks of starting the treatment, but normalization of muscle enzymes may take much longer.
Management of IBM. Patients with IBM usually present after several years of gradual muscle weakness. The older the patient's age at onset of IBM, the more rapid is the muscle loss. On average, patients lose muscle over a 10-year period before seeking medical attention. In contrast to other types of inflammatory myopathies, IBM is relatively resistant to standard immunosuppressive therapies. Corticosteroids alone appear to have a limited role in patients with IBM. Methotrexate and azathioprine alone or in combination also were shown at best to have a minor benefit. Adding IVIG to corticosteroids also did not clearly show any further benefit.11,12
The current recommendation for treatment of IBM focuses on physical and occupational therapy and orthotic devices. A tailored home exercise therapy 5 days a week was beneficial in one study.11 Currently, there are a few ongoing trials using CAMPATH-1, rituximab, and stem cell transplant for treatment of IBM.
Malignancy in Inflammatory Myopathies
The association between malignancy and autoimmune myopathies has been described and confirmed by multiple epidemiological studies. Malignancy can occur before, at the same time as, or after the diagnosis of myopathies. The close relationship between inflammatory myopathies and cancer is consistent with the concept that paraneoplastic processes linked to oncogenesis, and autoimmunity may play a significant role in the development of cancer.
A study from Sweden showed that 15% of patient with dermatomyositis and 9% of patients with polymyositis were diagnosed with some sort of malignancy in a 20-year period following diagnosis of myopathy. Dermatomyositis but not polymyositis was associated with increased risk of cancer mortality. Cancer incidence was higher in dermatomyositis and patients older than 65 years. The incidence of the diagnosis of cancer was the highest during the next 2 years following diagnosis of myopathy. Adenocarcinoma of the lung, ovaries, cervix, pancreas, and stomach comprised 70% of cancers associated with the myopathies.29
Summary
Dermatomyositis, polymyositis, IBM, and autoimmune necrotizing myositis are clinically, histologically, and pathogenically distinct categories of the idiopathic inflammatory myopathies. Dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy are responsive to immunosuppressive therapy, in contrast with IBM, which is generally refractory to currently available drugs. Major advances in treatment have improved the outcomes of patients with these diseases. There is much yet to accomplish especially in IBM, and the many complications of these diseases involving other organ systems including the lungs and the heart. Improvement is needed to reduce adverse outcomes of the medications used to treat these conditions. Greater understanding of the pathogenic bases of these disorders along with well-designed controlled trials using emerging new therapies can help improve the management of patients with these diseases.
Primary care physicians play an important role in optimizing care of these patients by recognizing these diseases in their early stages; ordering the preliminary studies including muscle enzymes, EMG, or MRI; and referring the patients to specialists. These patients often need proper cancer screening to rule out hidden malignancy. Collaboration of the primary care physician with the specialist can maximize management of these patients and minimize the side effects of corticosteroids and other immunotherapies.
References
1. Dimachkie M. Idiopathic inflammatory myopathy. J Neuroimmunol 2011;231:32-42.
2. Limaye VS, et al. Idiopathic inflammatory myopathies. Intern Med J 2009;39:179-190.
3. Phillips BA, et al. Prevalence of sporadic inclusion body myositis in Western Australia. Muscle Nerve 2000;23:970-972.
4. Firestein G, et al. Kelley's Textbook of Rheumatology. 8th ed. Inflammatory Disease of Muscle. 2009;78:1353.
5. Madan V, et al. Defining cancer risk in dermatomyositis. Part II. Assessing diagnostic usefulness of myositis serology. Clin Exper Dermatol 2009;34:561-565.
6. Bohan A, Peter JP. Polymyositis and dermatomyositis. N Engl J Med 1975;292;403-407.
7. Hodjendik J, Amato A. 119th ENMC international workshop. Trial design in adult inflammatory myopathies. Naarden, Netherland; 10-12 October 2003: 337-345.
8. Dalakas MC. Inflammatory muscle disease: A critical review on pathogenesis and therapies. Curr Opin Phamacol 2010;10:346-352.
9. Schmidt J, Dalakas MC. Pathomechanism of inflammatory myopathies: Recent advances and implications for diagnosis and therapies. Curr Opin Pharmacol 2010;4:241-250.
10. Targoff IN. Myositis specific autoantibodies. Curr Rheumatol Rep 2006;8:196-203.
11. Amato AA, Barohn RJ. Inclusion body myositis: Old and new concepts. J Neurol Neurosurg Psychiatry 2009;1186-1193.
12. Solorzano GE, Phillips LH 2nd. Inclusion body myositis: Diagnosis, pathogenesis, and treatment options. Rheum Dis Clinic North Am 2011;37:173-183.
13. van der Meulen MF, et al. Polymyositis: An overdiagnosed entity. Neurology 2003;61:316-321.
14. Grable-Esposito P, et al. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010;41:185-190.
15. Mosca M, et al. Undifferentiated connective tissue diseases (UCTD): A review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol 1999;17:615-620.
16. Bohan A, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine 1977;56:225-286.
17. Dalakas C. Review: An update on inflammatory and autoimmune myopathies. Neuropathol Appl Neurobiol 2011;37:226-242.
18. Gunawardena H, et al. Myositis-specific autoantibodies: Their clinical and pathogenic significance in disease expression. Rheumatology 2009;48:607-612.
19. Gunawardena H, et al. Newly identified autoantibodies: Relationship to idiopathic inflammatory myopathy subsets and pathogenesis. Curr Opin Rheumatol 2008;20:675-680.
20. Bromberg MB, Albers JW. Electromyography in idiopathic myopathies. Mt Sinai J Med 1988;55:559-464.
21. Park H, et al. Use of magnetic resonance imaging and P-31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum 1995;38:68-77.
22. Dorph C, et al. Percutaneous conchotome muscle biopsy. A useful diagnostic and assessment tool. J Rheumatol 2001;28:1591-1599.
23. Klippel J, Stone J. Primer on Rheumatic Diseases. 13th Ed. Idiopathic Inflammatory Myopathies. Pathology and Pathogenesis 2008;18:368-374.
24. Hengstman GJ, et al. Treatment of inflammatory myopathies: Update and practical recommendation. Expert Opin Phamacother 2009;10:1183-1190.
25. Joffe MM, et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993;94:379-387.
26. Isabelle M, Rouen C. Therapy of polymyositis and dermatomyositis. Quarterly Medical Review www.Sciencedirect.com; February 2011.
27. Majmudar S, et al. Treatment of adult inflammatory myositis with rituximab: An emerging therapy for refractory patients. J Clin Rheumatol 2009;15:338-340.
28. Dalakas MC, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993;329:1993-2000.
29. Sigurgeirsson B, et al. Risk of cancer in patients with dermatomyositis and polymyositis. A population-based study. N Engl J Med 1992;326:363-367.
The idiopathic inflammatory myopathies are a heterogeneous group of autoimmune syndromes characterized by subacute or chronic muscle weakness and skeletal muscle inflammation. Of the idiopathic inflammatory myopathies, the best recognized subsets are polymyositis, dermatomyositis, inclusion body myositis (IBM), and the newly described autoimmune necrotizing myopathy.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.