Cytotoxic Effects of Stress Induced Hyperglycemia
Special Feature
Cytotoxic Effects of Stress Induced Hyperglycemia
By Karen L. Johnson, PhD, RN
In 2001, the leuven study was the first randomized control trial that demonstrated the benefit of controlling hyperglycemia in non-diabetic critically ill surgical patients.1 Van den Berghe and colleagues demonstrated that intensive insulin therapy to maintain blood glucose at or below 110 mg/dL reduced mortality and morbidity (see Table 1). Krinsley compared the outcomes of 800 patients admitted consecutively to the ICU immediately before the onset of a glucose management protocol to those first 800 patients admitted after institution of the protocol.2 This more recent study reported similar findings in a heterogeneous population of non-diabetic critically ill adults: reduced hospital mortality 29.3% (P = .002), decreased ICU length of stay 10.8% (P = .01), reduction in renal insufficiency 75% (P = .03), and reduction in use of RBC transfusions 18.7% (P = .04).
Based on this emerging evidence, there are increased efforts around the world to maintain strict glycemic control in non-diabetic critically ill patients. Management of hyperglycemia through the use of insulin protocols is a new standard in critical care. Several protocols have been evaluated and reported in the literature.2-5
How could such a simple intervention, preventing hyperglycemia with insulin, prevent sepsis, MODS, and death? Why would preventing acute hyperglycemia improve mortality and morbidity in this patient population? What is the mechanism by which acute hyperglycemia causes so many complications? The answers to these questions are complex and largely speculative.
The purpose of this essay is to present some of the evidence currently available on the cytotoxic effects of stress induced hyperglycemia. Understanding the mechanisms through which stress hyperglycemia occurs and the role it plays in patient outcomes may enable clinicians to understand the importance of glycemic control in non-diabetics in the ICU. We will begin with a review of the etiology of hyperglycemia in this patient population.
Stress Induced Hypergylcemia
The initial physiologic response to acute stress (injury, illness) results in an increased availability of metabolic substrates for energy production: free fatty acids, amino acids, and glucose. This acute metabolic response is initiated by activation of the hypothalamic-pituitary-adrenal axis and the sympathetic nervous system. Excessive counterregulatory hormone release and overproduction of cytokines are the major factors responsible for stress hyperglycemia in the non-diabetic host.6 Epinephrine and norepinephrine via adrenergic activity directly inhibit insulin secretion. Epinephrine decreases hepatic glycogen synthesis, increases glycogenolysis, increases hepatic gluconeogenesis and increases skeletal muscle insulin resistance by altering post-receptor signals. Cortisol increases gluconeogenesis and contributes to skeletal muscle insulin resistance.
Insulin resistance is defined as the existence of metabolic characteristics of insulin deficiency (protein catabolism, hyperglycemia, lipolysis) despite adequate plasma insulin concentration.7 The mechanism underlying the inability of hyperinsulinemia to suppress hepatic gluconeogenesis is unknown. However, skeletal muscle is the major site of reduced insulin-mediated glucose disposal and this contributes to whole body insulin resistance in critical illness.8,9 Overall, stress hyperglycemia results from a combination of enhanced hepatic glucose production and decreased peripheral glucose utilization via insulin resistance.
Prior to the late 1990’s, there was little argument for medical interference with this normal physiologic process during the first few hours or days of critical illness or trauma.10 In the past several years, however, an increasing amount of evidence suggested otherwise.
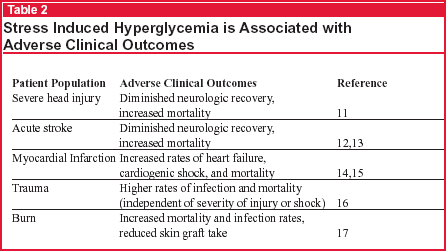
Stress hyperglycemia, defined as a blood glucose greater than 200 mg/dL, is most often evident shortly after admission to the ICU.6 It has been associated with adverse clinical outcomes in a variety of critically ill patients including those with closed head injury, stroke, myocardial infarction, trauma, and burns (see Table 2, below). As demonstrated in the Leuven1 and Kinsley2 studies, these complications decreased with normoglycemia induced with insulin. These results prompt the question, Are the benefits brought about directly by the infused insulin per se, or by the prevention of hyperglycemia?
In an effort to answer this question, Van den Berghe et al returned to their original data to examine the factors determining insulin requirements and the impact of insulin dose versus blood glucose control on the observed outcome benefits.18 Mulivariate logistic regression analysis indicated that it was actually the lowering of blood glucose (rather than amount of insulin) that was associated with the reduction in mortality (P < .001), critical illness polyneuropathy (P < .001), bacteremia (P = .02), and inflammation (P = .0006).
Why is acute hyperglycemia so toxic in critically ill patients when it takes years for hyperglycemia to cause disorders in diabetic patients? How is acute hyperglycemia toxic to cells? The effects of acute hyperglycemia appear to cause both intra- and extracellular pathophysiologic processes.
Intracellular Cytotoxic Effects of Stress-Induced Hyperglycemia
A recent review suggests that intracellular cytoxic effects of stress induced hyperglycemia maybe due to mitochondrial dysfunction as a result of accentuated intracellular glucose overload and more pronounced toxic side effects of glycolysis and oxidative phosphorylation.19 A quick review of cellular glucose uptake helps to put this explanation into context.
Normal Cellular Uptake of Glucose
Skeletal muscle is the major site of peripheral insulin mediated glucose uptake in humans, accounting for 80% of whole body glucose disposal under hyperinsulinemic-euglycemic clamp conditions.20 Stimulation of the movement of glucose into skeletal muscle is a critical component of the physiologic response to insulin. The insulin receptor is a transmembrane heterodimeric protein receptor. When insulin binds with it, insulin receptor substrate molecules phosphorylate. From there, intracellular signaling pathways culminate in the translocation of intracellular glucose transporters to the cell membrane. The GLUT transporters act as vesicles to transport glucose inside the cell. In skeletal muscle, this involves translocation of GLUT-4 and GLUT-1 vesicles. GLUT-4 transporters are insulin dependent and can become activated and increase glucose uptake several fold. GLUT-1 transporters ensure basal glucose uptake into cells. GLUT-1 transporters downregulate in normal cells when exposed to hyperglycemic conditions to protect the cell. Upon insulin removal, the transporters return to their intracellular pool.
Uptake of glucose by skeletal muscle cells is dependent on the GLUT-4 insulin-dependent transporters. Other cells take up glucose independently of insulin. Cellular compartments that take up glucose independently of insulin include the central and peripheral nervous system, hepatocytes, and endothelial, epithelial, and immune cells.19 GLUT-1, GLUT-2, and GLUT-3 transporters facilitate insulin independent glucose transport into these tissues (see Table 3).
Once inside the cell, glucose is phosphorylated into either glycogen for storage or glycolysis, which leads to its use in the Krebs Cycle resulting in energy production via oxidative phosphorylation. In the presence of oxygen, aerobic metabolism ensues. Glucose undergoes glycolysis where pyruvate is transformed to acetyl coenzyme and oxidative phosphorylation generates adenosine triphosphate (ATP). A small amount of superoxide is produced, but it is detoxified by manganese superoxide dismutase.
Hyperglycemia Induced Mitochondrial Dysfunction
Angiogensin II, hypoxia, cytokines, and endothelin-I have been shown to upregulate expression and membrane localization of GLUT-1 and GLUT-3 transporters.21-24 When high levels of these substances are present, as they frequently are in critically ill patients, more glucose enters cells.
As more glucose enters cells and aerobic metabolism occurs, more superoxide is produced. It reacts with nitric oxide to form peroxynitrite. Peroxynitrite breakdown intracellular proteins, most notably mitochondria. The formation of peroxynitrite is compounded in critical illness in the presence of iNOS and additional superoxide production from hypoxia/reperfusion.25 Therefore, it appears that intracellular hyperglycemia induces mitochondrial dysfunction in those cells that take up glucose independent of insulin (see Table 3).
Postmortem liver biopsies performed in the Leuven study seem to support this mechanism. Hepatocytes use GLUT-1, GLUT-2, and GLUT-3 transporters facilitate insulin independent glucose uptake. Liver biopsies revealed profound abnormalities in hepatocyte mitochondria in patients who received conventional insulin therapy but no major abnormalities in patients who remained normoglycemic.
Controlling hyperglycemia may prevent mitochondrial dysfunction in cells that allow glucose to enter passively and may explain some of the results of reported in the Leuven study.1 (see Table 4, below). Further investigation of the mitochondrial abnormalities in tissues that take up glucose passively should be investigated in greater detail.

Extracellular Cytoxic Effects of Stress Hyperglycemia
Intracellular hyperglycemia induced mitochondrial dysfunction only explains some of the pathophysiology of stress induced hyperglycemia. While increased intracellular glucose occurs in some cells, there appears to be decreased glucose uptake by other cells, particularly in skeletal muscle cells. This results in extracellular hyperglycemia.
Hyperglycemia, commonly associated with septic shock, may be the result of insulin resistance in skeletal muscle cells. Experimental data from rats indicate that LPS alters multiple steps in the insulin signal transduction pathway.26 Glucocorticoids impair insulin mediated glucose uptake in skeletal muscle likely by inhibiting translocation of the GLUT-4 transporter.27 Defects of GLUT-4 may be the underlying mechanism of peripheral insulin resistance in critical illness.28 TNF-a produces insulin resistance in both liver and skeletal muscle through modification of signaling properties of insulin receptor substrates.29 Interestingly, prolonged bedrest (seven days) produced insulin resistance in skeletal muscle in healthy subjects.30 Catecholamines may also play a role in insulin resistance. Blockade of beta-2 adrenergic receptors prevents the decline in insulin-mediated glucose uptake in septic rats.31
Overall the mechanisms by which insulin signal transduction pathways and/or GLUT4 expression and function may be altered leading to reduced insulin-stimulated glucose disposal in critical illness are far from established. But the net result is increased extracellular glucose.
Many theories have been postulated and there is extensive research in an effort to explain the pathogenesis of prolonged hyperglycemia and its effect on biochemical changes in cells, but less is known about the effects of short term acute hyperglycemia. Short term hyperglycemia increases apoptosis in cultured endothelial cells.32,33 After several days of exposure to hyperglycemic conditions, endothelial cells have increased oxidative stress, increased intracellular calcium, loss of mitochondrial polarization, and decreased intracellular ATP content.34 Acute hyperglycemia decreases respiratory burst of alveolar macrophages and influences phagocytic cells,35 impairs immune function by altering cytokine production from macrophages and decreases lymphocyte proliferation.36 Acute hyperglycemia increases release of IL-6 and TNF—which suggests a pivotal role of hyperglycemia in the inflammatory responses in sepsis.37
Summary
Stress-induced hyperglycemia, associated with adverse clinical outcomes, is present within hours of ICU admission and results from a combination of enhanced glucose production, increased glucose uptake by some cells, and decreased peripheral glucose uptake by skeletal muscle cells (via insulin resistance). Increased glucose uptake produces mitochondrial dysfunction as a result of accentuated intracellular glucose overload and more pronounced toxic side effects of glycolysis and oxidative phosphorylation. Peroxynitrite forms and breaks down intracellular proteins, most notably the mitochondria. The net result is mitochondrial dysfunction. Insulin resistance results in decreased glucose uptake by skeletal muscle cells with the net result of increased extracellular glucose concentrations. Exposure to hyperglycemic conditions has negative effects on cells particularly endothelial cells and immune cells and also potentates cytokine release.
Controlling stress-induced hyperglycemia with insulin in non-diabetic critically ill patients decreases morbidity and mortality. The intracellular and extracelluar effects of stress-induced hyperglycemia have been elucidated in an effort to help clinicians better understand why prevention of stress induced hyperglycemia via insulin protocols is so important in decreasing the morbidity and mortality of critical illness.
Karen Johnson, PhD, RN, School of Nursing University of Maryland, is Associate Editor for Critical Care Alert
References
1. Van den Berghe G, et al. N Engl J Med. 2001;345: 1359-1367.
2. Krinsley JS. Mayo Clin Proc. 2004;79:992-1000.
3. Brown C, Dodek P. Crit Care Med. 2001;29:1714-1719.
4. Van den Berghe G. Int Diab Monitor. 2002;14(6):1-6.
5. Goldberg PA, et al. Diab Care. 2004;27:461-467.
6. McCowen KC, et al. Crit Care Clin. 2001;17:107-124.
7. Carlson GL. Br J Surg. 2003;90:259-260.
8. Lang CH, et al. Metabolism. 1990;39:1096-1107.
9. Robinson LE, van Soeren MH. AACN Clin Issues. 2004;15:45-62.
10. Van den Berghe G, et al. J Clin Endocr Metab. 1998; 83:1827-1834.
11. Rovlias A, Lotsou S. Neurosurgery. 1999;46:335-342.
12. Parsons MW, et al. Ann Neurol. 2002;52:20-28.
13. Williams LS, et al. Neurology. 2002;59:67-71.
14. Malmberg K, et al. Circulation. 1999;99:2626-2632.
15. Capes SE, et al. Lancet. 2000;355:773-778.
16. Laird AM, et al. J Trauma. 2004;56:1058-1062.
17. Gore DC, et al. J Trauma. 2001;51:540-543.
18. Van den Berg G, et al. Crit Care Med. 2003;31:359-366.
19. Van den Berghe G. J Clin Invest. 2004;114:1187-1195.
20. Kruszynska YT, Olefsky JM. J Investig Med. 1996;44: 413-428.
21. Quinn LA, McCumbee WD. J Cell Physiol. 1998;177: 94-102.
22. Clerici C, Matthay MA. J Appl Physiol. 2000;88: 1890-1896.
23. Shikhman AR, et al. J Immunol. 2001;167:7001-7008.
24. Sanchez-Alvarez R, et al. J Neurochem. 2004;89: 703-714.
25. Aulak KS, et al. Am J Physiol Heart Circ Physiol. 2004;286:H30-H38.
26. Fan J, et al. Shock. 1996;6:164-170.
27. Dimitriadis G, et al. Biochem J. 1997;321(Pt 3): 707-712.
28. Fink RI, et al. Metabolism. 1992;41:867-902.
29. Hotamislingil GS, Spiegelman BM. Diabetes. 1994;43: 1271-1280.
30. Stuart CA, et al. Metab: Clinical & Experimental. 1988;37:802-806.
31. Lang CH. Am J Physiol. 1992;263:E703-E711.
32. Baumgartner-Parzer SM, et al. World J Gastroenterol, 2003; 9:1824-1827.
33. Ho FM, et al. Circulation. 2000;101:2618-1624.
34. Ido Y, et al. Diabetes. 2002;51:159-167.
35. Kwoun MO, et al. JPEN. 1999;21:91-95.
36. Losser MR, et al. J Appl Physiol. 1997;83:1566-1574.
37. Wen-Kui Y, et al. World J Gastroenterol. 2003;9:1824-1827.
Based on emerging evidence, there are increased efforts around the world to maintain strict glycemic control in non-diabetic critically ill patients. Management of hyperglycemia through the use of insulin protocols is a new standard in critical care. Several protocols have been evaluated and reported in the literature.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.