Thrombocytopenia
Thrombocytopenia
Author: Jonathan Glauser, MD, FACEP, Department of Emergency Medicine, Cleveland Clinic, Cleveland, OH.
Peer Reviewers: Catherine A. Marco, MD, FACEP, Professor, Department of Surgery, Division of Emergency Medicine, University of Toledo College of Medicine, Toledo, OH; and Steven M. Winograd, MD, FACEP, Attending Physician, Emergency Medicine, Horton Hill Hospital, Middleton, NY, Arden Hill Hospital, Goshen, NY.
Most cases of thrombocytopenia seen in the emergency department (ED) are expected. Patients are known to have hematological disease or are receiving chemotherapy. At times, however, the ED physician is confronted with an unexpected laboratory finding in an assymptomatic patient, or with a patient who is bleeding. The challenge, as usual, is to determine the need for acute treatment and the appropriate disposition.
As this article discusses, one of the most important diagnostic procedures in patients with thrombocytopenia is the peripheral blood smear. The simple test will often determine the presence of pseudothrombocytopenia and more serious causes such as thrombotic thrombocytopenia and hemolytic uremic syndrome.
Sandra M. Schneider, MD, FACEP, Editor
Introduction
Case 1. A 45-year-old patient presents to the emergency department (ED). He has a long history of alcohol abuse, and is clearly intoxicated this evening. While you study his records and perform a medical screening examination, you learn that he has a diagnosis of cirrhosis. The only medication he is prescribed is folate, but he has been non-compliant with this for years. His examination is unremarkable except for splenomegaly. Specifically, he is alert without petechiae or ecchymoses. His urinalysis is negative for blood, and his stool is guaiac negative. His laboratory values are remarkable for a hemoglobin of 13.4 grams, a normal white blood count and differential, an INR of 1.0, normal electrolytes, and a platelet count of 8000/µL.
Case 2. A 3-year-old child presents with complaint of reddish spots on his legs of 3-4 days duration. He has been in good health, although his mother reports an upper respiratory infection one week ago, for which the child did not receive medication. On examination, his vital signs are normal, and he shows no evidence of recent epistaxis, and no blood in his urine or stool. The reddish spots do not blanch and are 2-3 mm in diameter. A complete blood count reveals a hemoglobin of 13.9, with a white blood count of 5,000 and a platelet count of 19,000/µL.
Case 3. A 56-year-old woman presents to a teaching hospital with shortness of breath of sudden onset. She has recently been hospitalized for a deep venous thrombosis in her left leg. Initially she had been fully heparinized with unfractionated heparin (UFH), then discharged from the hospital 4 days later with a therapeutic INR, on warfarin. She insists that she has been compliant with her warfarin therapy, and her measured INR is 2.7. Her hemoglobin is 14.3, but her platelet count is only 22,300/µL. Her story is typical for pulmonary embolism, and the primary physician wants to re-start heparin and admit her to the hospital for a filter placement. The intern is skeptical that she could have developed a pulmonary embolism given that her INR is therapeutic, her platelet count is very low, and therefore she should not be at risk to have a clot.
Thrombocytopenia
The normal platelet count in adults ranges from 150,000 to 450,000/microliter (µL). Thrombocytopenia is defined as a platelet count of less than 150,000/µL (150 x 109/L), with the implication that 2.5% of the normal population will have a platelet count lower than this. Clinically, thrombocytopenia may be asymptomatic or may present with bleeding, petechiae, or easy bruising. Regardless of cause, thrombocytopenia stimulates an increase in free thrombopoietin, which attempts to increase platelet production. Young platelets are larger than mature platelets and are often seen on the blood smear. Broadly, low platelet counts may be caused by decreased production, increased destruction, splenic sequestration, or a combination of the three. Approximately one-third of platelets are sequestered in the spleen, and the other two-thirds circulate for 7-10 days. The cause for a low platelet count may not be immediately apparent, especially in patients without evidence for systemic disease. A major goal in the ED is to control bleeding if present (often requiring platelet transfusion), and assess the risk of bleeding. The risk of spontaneous bleeding increases when the platelet level falls below 10,000 and 20,000/µL
The cause for the thrombocytopenia may not be immediately apparent, especially in patients without evidence of systemic disease. In many cases the exact cause can be determined later. However there are several disorders that should be determined in the ED such as thrombotic thrombocytopenia purpura, hemolytic uremic syndrome, and heparin-induced thrombocytopenia.
Pseudothrombocytopenia
The platelet count can be falsely low for a number of reasons. First, if the blood sample is inadequately anticoagulated, platelet clumps might be recognized as leukocytes by cell counters. A visual check of a blood smear generally will determine this diagnosis. The white blood cell count might be falsely elevated by up to 10% in this case.1
Some patients have intrinsic EDTA-dependent agglutinins, which lead to spurious leukocytosis and thrombocytopenia. Similar to inadequately anticoagulated blood, EDTA-induced platelet clumping can be diagnosed by examination of the peripheral smear. If the peripheral smear shows platelet clumping, the platelet count should be repeated using heparin or sodium citrate as the anticoagulant, correcting the platelet count for dilution if citrate solution is used.2
Dilutional thrombocytopenia occurs in patients who have sustained massive blood loss with subsequent transfusion of packed red blood cells (RBCs). In patients receiving 20 units of packed red blood cells over 24 hours, platelet counts have been reported to be 25,000 to 61,000/µL.3 This may be prevented by giving platelet concentrate transfusions to patients receiving more than 20 units of packed RBCs over a 24 hour period.2
Dilutional thrombocytopenia also may be caused by splenomegaly. Normally, approximately one-third of circulating platelets are sequestered in the spleen, where they are in equilibrium with circulating platelets. Patients with cirrhosis, portal hypertension, and splenomegaly may have apparent thrombocytopenia, as splenic sequestration of platelets can be increased to as high as 90% with portal hypertension. However, since the total platelet mass and survival is relatively normal, clinical bleeding is rarely a problem from low platelets due to splenomegaly alone.4
Categories and Causes for Thrombocytopenia
Platelet counts may be depressed because of decreased production or due to increased platelet destruction.
Decreased platelet production by the bone marrow may be related to viral infections: mumps, rubella, varicella, hepatitis C,5 or Epstein-Barr virus. Neonatal infections such as cytomegalovirus (CMV) or rubella may present with other findings in addition to thrombocytopenia. The human immunodeficiency virus may damage megakaryocytes directly. Thrombocytopenia also may be related to measles vaccination.6 A decreased platelet count due to viral infection may be more severe if present in a patient with bone marrow suppression, such as from chemotherapy. Chemotherapy or radiation therapy may decrease platelet production as well as other blood cells. Marrow infiltration from lymphoma or leukemia or myelofibrosis may cause deficits in multiple cell lines. Some congenital or acquired disorders are associated with bone marrow aplasia or hypoplasia. Alcohol may suppress platelet production, as may vitamin B12 or folate deficiency. Decreased platelet production may be drug-related, as with heparin, quinidine, valproic acid, or quinine. (See Table 1.)
 |
Increased platelet destruction may be due to immune mechanisms, such as systemic lupus erythematosus (SLE), antiphospholipid syndrome, or idiopathic thrombocytopenic purpura (ITP). With ongoing platelet destruction, the circulating young platelets are usually large, indicating that the bone marrow is producing new/larger platelets to compensate for increased destruction. Thrombocytopenia may be related to consumption, as in disseminated intravascular coagulation (DIC) or to alloimmune destruction, as may occur in neonates, post-transplantation, or post-transfusion states. Some viral infections have been related to platelet destruction, notably infectious mononucleosis, cytomegalovirus, and HIV. Fever and thrombocytopenia in an adult or child with a history of recent travel should suggest malaria, and a blood smear examined for parasites.7 Thrombotic thrombocytopenic purpura–hemolytic uremic syndrome (TTP–HUS), and the HELLP syndrome of hemolytic anemia, elevated liver function tests, and low platelet count are also associated with increased platelet destruction. (See Table 1.)
Incidental thrombocytopenia of pregnancy, or gestational thrombocytopenia, is generally mild, asymptomatic, and resolves spontaneously rapidly postpartum. It tends to occur in late gestation and is not associated with fetal thrombocytopenia. Platelet counts are generally greater than 70,000/µL.2
Splenic sequestration may be suspected in any patient with splenomegaly. An estimate of splenic size, therefore, should be made based upon physical examination supplemented if necessary by ultrasound or computed tomography. Portal hypertension secondary to liver disease, or splenic infiltration with myeloproliferative or lymphoproliferative disorders are common causes of splenomegaly.
There are unusual causes of thrombocytopenia in children, some congenital. Thrombocytopenia associated with giant hemangioma of infancy is called the Kasabach-Merritt syndrome. Alloimmunization secondary to incompatibility of platelet antigens between mother and fetus is termed neonatal alloimmune thrombocytopenia (NAIT), due to platelet destruction by maternal IgG alloantibodies that have crossed the placenta. More common is incompatibility of the HPA-1a or PIA1 antigen, causing thrombocytopenia, and possibly hydrocephalus, seizures, hyperbilirubinemia, and fetal loss. Treatment is with PIA1-negative platelet transfusion.
Mild thrombocytopenia is suggestive of Bernard-Soulier syndrome, a congenital syndrome associated with platelet dysfunction and absence of glycoprotein 1b, V, and IX.8 These proteins serve as receptors for von Willebrand factor and their absence results in decreased platelet adhesion.
Congenital idiopathic amegakaryocytic thrombocytopenia presents in infants as isolated thrombocytopenia, although this may progress later to pancytopenia.9 Hereditary amegakaryocytopoiesis usually is associated with skeletal abnormalities.
Thrombocytopenia with absent radii (TAR) syndrome is one associated with radial agenesis and other upper limb defects.10 Wiskott-Aldrich syndrome is associated with small platelets, thrombocytopenia, eczema, and immunodeficiency. Hematopoietic cell transplant may be curative.11
The Clinical Presentation of Thrombocytopenia
Asymptomatic. Patients with thrombocytopenia may be asymptomatic, with detection only on a complete blood count. Mucosal bleeding may present as epistaxis or gingival bleeding. Cutaneous bleeding may manifest itself as petechiae, bullous hemorrhages, or ecchymoses. Petechiae are red, flat, non-palpable, discrete lesions that are pinhead sized and tend to cluster on the feet and ankles. (See Figure 1.) They are nontender and are due to the presence of extravasated red blood cells. Ecchymoses are also nontender areas of bleeding into the skin from extravasated blood, and usually develop without noticeable trauma. Hemoptysis, hematuria, and hematochezia may be presenting signs.
 |
Prolonged bleeding during menstruation (menorrhagia) or bleeding between menses (metrorrhagia) are common presentations. Patients with platelet abnormalities tend to bleed immediately after vascular trauma, as opposed to delayed bleeding characteristic of coagulation disorders such as hemophilia. Bleeding into the central nervous system is uncommon without preceding trauma, but is the most common cause of death due to thrombocytopenia.2 In patients with coagulation disorders such as hemophilia, bleeding tends to be deeperin the tissues, muscles, and joints, as well as more delayed bleeding and more post-surgical bleeding.
Bleeding Risks. The risks of bleeding at any given platelet level may depend upon the underlying cause for the thrombocytopenia. Surgical bleeding due solely to a reduction in the number of platelets is unlikely unless the platelet count is less than 50,000/µL. Spontaneous bleeding does not occur until the platelet count is less than 10,000 to 20,000/µL. On the other hand, patients with idiopathic thrombocytopenic purpura (ITP) do not usually have serious bleeding, suggesting that the young platelets found in these patients are more hemostatically active than mixed age or older platelets in normal subjects. In patients with ITP, severe life-threatening bleeding may not occur until platelet counts are less than 6,000/µL, while self-limiting and spontaneous bleeding did not occur until platelet counts were less than 40,000/µL and 12,000/µL respectively, in one report.12 (See Table 2.)
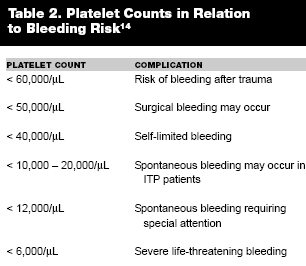 |
The risk of lumbar puncture in the thrombocytopenic patient is of importance in the ED. One study reported 29 lumbar punctures in children with platelet counts less than 10 x 109/L or less, and another 170 with platelet counts of 11-20 x 109/L, with no complications, among children with acute lymphoblastic leukemia. The authors felt that prophylactic platelet transfusions were not definitely indicated prior to lumbar puncture in thrombocytopenic children.13
The History in the Thrombocytopenic Patient
A history of recent viral or rickettsial infection, or of recent travel, should be sought even in a patient with known bone marrow suppression. A history of recent live virus vaccination and medication exposure or a positive family history of bleeding or thrombocytopenia should be sought. Poor nutritional status, including alcoholism or history of certain medications, may give a clue as to etiology. Dietary habits or antibiotic use that might predispose to deficiencies of vitamin K, vitamin B12, and folic acid should be queried in patients with suspected bleeding disorder. While a careful drug history is paramount, thrombocytopenia caused by undisclosed drug use has been described.14 Non-hematologic disorders known to decrease platelet counts, such as eclampsia, sepsis, DIC, hypothermia, or massive transfusions, may be evident. Sepsis is the most common cause for thrombocytopenia developing for the first time in a patient in an intensive care unit setting (up to 50% of the cases).15
The Physical Examination of the Thrombocytopenic Patient
Examination for lymphadenopathy and splenomegaly may indicate the presence of a disseminated disorderinfectious or malignant. Funduscopic examination may reveal evidence for hemorrhage, as CNS bleeding is the most common cause for death in the thrombocytopenic patient. Examination of the stool may reveal occult blood. Skin evaluation and examination of catheter, drain, or surgical sites may reveal evidence for bleeding. The size of the spleen should be assessed as splenomegaly is suggestive of lymphoma, malaria, mononucleosis, or other disorder associated with splenic sequestration.
The Evaluation of the Patient with Thrombocytopenia. Laboratory evaluation of the thrombocytopenic patient begins with a complete blood count and examination of the peripheral smear for estimation of the morphology and number of platelets, presence of platelet clumping, and morphology of platelets. Any associated white and red blood cell changes should be noted as well.
A peripheral smear of non-anticoagulated blood should be examined. As noted earlier, the presence of platelet clumps may result in a spuriously low platelet count due to inadequate sample anti-coagulation or to EDTA. Repeat blood sampling with an alternate anticoagulant such as heparin or sodium citrate helps to make the diagnosis. The blood smear is also important to examine to diagnose some of the more severe syndromes of TTP, HUS, and DIC.
A number of congenital thrombocytopenic disorders manifest on the peripheral smear as alterations in size, or with abnormal platelet granules, or with neutrophilic inclusions.16 The May-Hegglin anomaly is an autosomal dominant trait characterized by giant platelets, leukocyte inclusion bodies, and mild to moderate thrombocytopenia. The laboratory particle counter may not recognize the platelets and give a falsely low reading; patients are generally asymptomatic, but may have a mild bleeding disorder. Alport's syndrome of congenital nephritis may present with hematuria and progressive renal failure. Some families have thrombocytopenia and deafness. Peripheral smear may show leukocyte inclusion bodies. Wiskott-Aldrich syndrome is an X-linked disorder characterized by immune deficiency, eczema, and thrombocytopenia with small platelets.
Inadequate sample anticoagulation may reveal EDTA-induced platelet clumping in the peripheral smear, as noted earlier. Repeat blood sampling with a different anticoagulant such as heparin or sodium citrate confirms the diagnosis.
The cause for decreased platelet production may be evident on smear. Circulating blast cells indicate the presence of acute leukemia. A leukoerythroblastic blood picture with nucleated red blood cells and early myeloid forms in the blood suggests bone marrow invasion with tumor, fibrosis, or granulomatous infection such as tuberculosis. Other cytopenias may be present in myelodysplastic states. Large oval red blood cells with hypersegmentation of neutrophils are present on vitamin B12 or folate deficiency.2
Increased platelet destruction may be suggested with a microangiopathic blood picture, with fragmented red blood cells, hemolytic anemia, and elevated lactate dehydrogenase. These findings occur in disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura and hemolytic uremic syndrome (TTP-HUS).
Bone marrow aspiration and biopsy is indicated on nearly all patients with thrombocytopenia severe enough to constitute a risk for major bleeding. An exception is the patient younger than age 60 with isolated thrombocytopenia and no evidence for underlying disorder with a presumptive diagnosis of idiopathic thrombocytopenic purpura (ITP, see below).17 Even if the diagnosis of ITP is contemplated, it has been proposed that bone marrow examination be accomplished prior to starting steroid therapy, as this may confound the diagnosis of acute leukemia in children.18 The presence of normal to increased numbers of megakaryocytes indicates that the patient's thrombocytopenia is due, at least in part, to increased peripheral destruction, as in ITP. Decreased numbers of megakaryocytes along with overall decreased cellularity is consistent with decreased bone marrow production, as in aplastic anemia. Megaloblastic changes in the red blood cell and granulocytic series suggest vitamin B12 or folate deficiency. Dysplastic changes in the red cell, granulocytic, and megakaryocytic lineages suggest a myelodysplastic disorder.
Bone marrow invasion is suggested by the presence of malignant cells, granulomata, or collagen fibrosis. Rarely, there may be severe reduction or absence of megakaryocytes with no other abnormalities, suggesting the presence of an autoantibody against the thrombopoietin receptor, as in systemic lupus erythematosus.2
Drug-Induced Thrombocytopenia
The diagnosis of drug-induced thrombocytopenia can be made only by demonstrating resolution of thrombocytopenia once a suspected drug is discontinued.19 It is important to note that many patients with drug-induced thrombocytopenia are initially diagnosed as having idiopathic thrombocytopenic purpura. A cause and effect relationship depends upon establishment of the following:
- • Therapy with the suspected drug preceded the thrombocytopenia;
- • Recovery from the thrombocytopenia was complete and sustained after discontinuation of the drug;
- • Other drugs were continued or re-introduced after discontinuation of therapy with the suspected drug, with a sustained normal platelet countor the suspected drug was the only one used before the onset of thrombocytopenia;
- • Other causes for thrombocytopenia were excluded;
- • Re-exposure to the suspected drug resulted in recurrent thrombocytopenia.20
The number of medications associated with thrombocytopenia is quite long; one list from Denmark of thrombocytopenia-inducing agents included 110 different drugs.21 Some drugs, such as aspirin or non-steroidal anti-inflammatory drugs (NSAIDs), can exacerbate an underlying platelet disorder. Commonly cited agents include gold salts, heparin, valproic acid, phenytoin, indomethacin, sulfonamides, glycoprotein IIb/IIIa inhibitors, quinidine, and quinine. (See Table 3.) Heparin-induced thrombocytopenia is the most common drug-related cause of a drop in platelet count and is addressed separately.
 |
Some cytotoxic agents such as cisplatin and cyclophosphamide cause generalized bone marrow depression.22 Other drugs induce selective suppression of megakaryocyte production (thiazides, ethanol, tolbutamide). Some accelerate platelet destruction by immunologic (acetaminophen, methicillin, rifampin, procainamide, interferons alfa and beta, others) or non-immunologic (ristocetin, protamine sulfate) mechanisms.
Current laboratory assays for drug-dependent antiplatelet antibodies are not useful for diagnosing drug-induced thrombocytopenia in the ED.19 In some patients with a history that is typical for drug-induced thrombocytopenia, antibody tests may be negative.22 One reason for this may be that a drug metabolite produced in vivo is the actual sensitizing agent.23 Gold-induced immune thrombocytopenia may persist for months due to presence of the antigen in the reticuloendothelial system, but for the most part, thrombocytopenia should resolve within 2-4 weeks of discontinuation of an offending drug. Typically, a patient will have taken the sensitizing drug for approximately one week or intermittently over a longer period before presenting with petechiae or ecchymoses, but occasionally symptoms develop within 1 to 2 days of exposure to a drug. This is true particularly of the platelet inhibitor abciximab.24 Abciximab-induced thrombocytopenia may occur within 30 minutes to several hours after initial administration. Other types of drug-induced thrombocytopenia require a much longer period of drug administration to induce sensitization.25,26 Certain drugs that are cleared from body storage depots very slowly, such as phenytoin, may induce more prolonged thrombocytopenia.
Many patients with drug-induced thrombocytopenia have only petechial hemorrhages and occasional ecchymoses and require no treatment except to discontinue the sensitizing medication. More aggressive therapy including platelet transfusions may be necessary to prevent intrapulmonary or intracranial hemorrhage. Otherwise, treatment may include oral or intravenous steroids and intravenous immune globulin. The platelet count should be monitored frequently in patients receiving glycoprotein IIb/IIIa inhibitors, especially abciximab.27 Three patients in one series underwent splenectomy before the drug-induced (quinidine) etiology was discovered.28
Medication exposure may not be obvious and, at times, some detective work is necessary. Heparin-induced thrombocytopenia is discussed separately, although it is worth noting that heparin flushes of vascular access lines may not be noted separately in the medical record. Vancomycin mixed into joint replacement cement may not be known to the patient. Drug-induced thrombocytopenia has occurred due to herbal remedies as well.29
Idiopathic Thrombocytopenic Purpura
Idiopathic thrombocytic purpura (ITP) is an acquired auto-immune disease that results in the destruction of platelets. There is no definitive test for ITP, making this a diagnosis of exclusion. It is characterized by thrombocytopenia, with petechiae or purpura, and a normal bone marrow. ITP may occur in all age groups, and may present acutely or chronically. Acute ITP is more common in younger children and typically resolves within 1-2 months.
Immune thrombocytopenic purpura is classified as either primary or as secondary to an underlying disorder. Primary immune thrombocytopenia is the classic ITP that is not secondary to any systemic illness.30 Secondary immune thrombocytopenic purpura is associated with SLE, chronic lymphocytic leukemia, HIV infection, hepatitis C infection, and a variety of other disorders including therapy with drugs such as heparin or quinidine.31
ITP in children usually occurs between the ages of 2 and 4 years. There may be a history of antecedent viral illness, such as an upper respiratory infection or exanthem, but the child with ITP is otherwise in excellent health.32 The onset of petechiae, bruising, epistaxis, hematuria, or gastrointestinal hemorrhage is what brings the patient to medical attention. In approximately 90% of children with ITP, the disease is acute and self-limited, resolving within six months. Chronic ITP is more common in children older than age 10 years or younger than 1 year of age.33 Mortality is almost always associated with intracranial hemorrhage.34
With immune thrombocytopenias, platelet destruction is mediated by the production of autoantibodies that attach to circulating platelets. The initial inciting event in ITP is unknown, but mechanisms that lead to platelet destruction involve the activation of helper T cells and cytotoxic T cells, and antibodies against glycoproteins in the platelet membrane.31 Platelets coated with IgG antibodies undergo accelerated clearance through Fcγ receptors that are expressed through tissue macrophages. The antibody-coated platelets are removed by the reticuloendothelial system. The platelets that are not destroyed do function normally, and frequently bleeding is not a significant problem despite low platelet counts. The methods that are currently used to treat immune thrombocytopenic purpura are directed at different aspects of the cycle of antibody production and platelet sensitization, clearance, and production.
Acute ITP is unusual in adults. Most adults present with an indolent form of thrombocytopenia that may persist for many years, and is referred to as chronic ITP. Chronic ITP is more insidious, and approximately twice as many women as men are affected, most commonly between the ages of 18 and 40 years. It is more likely to occur in patients with underlying disease or immune disorder including systemic lupus erythematosus, Graves' disease, Hashimoto's thyroiditis, HIV, or antiphospholipid syndrome.
The CBC should demonstrate normal cell lines except for platelets. The peripheral smear is expected to show large platelets, but fewer platelets than normal. Platelet-associated IgG can be measured, but the specificity of the test is not high enough to make it clinically useful in making the diagnosis. The direct assay for the measurement of platelet-bound antibodies has an estimated sensitivity of 49-66% and an estimated specificity of 78-92%.35,36
Treatment of ITP includes avoidance of antiplatelet medications such as aspirin or non-steroidal anti-inflammatory drugs (NSAIDs). Fall risks should be addressed. If platelet counts are greater than 50,000/µL, no treatment is recommended. Otherwise, treatment should be initiated with steroids: 1-1.5 mg/kg or 60 mg of prednisone/day typically. A response rate of 50-80% can be expected, although when treatment is stopped, remission is sustained in only 10-30% of cases.31 If this does not elicit a response within 6-8 weeks, splenectomy may produce long-standing remission. Splenectomy may impair the clearance of antibody-coated platelets by the Fcγ receptors and may also impair the T cell/B cell interactions involved in antibody synthesis in some patients.30 The risks associated with splenectomy are small, but patients post-operatively have a lifelong risk of bacterial sepsis. For this reason, the American Society of Hematology recommends that splenectomy be considered in children who have had ITP for at least one year with symptomatic severe thrombocytopenia.17 Prior to splenectomy, patients should be immunized with pneumococcal and Hemophilus influenzae type B vaccines. Occasionally, patients may fail to respond to therapy because of the presence of an accessory spleen, which may be detectable on nuclear scanning.
For patients who decline splenectomy, a variety of third-line therapies are available. Plasmapheresis transiently removes autoantibody from the plasma. Rituximab, a monoclonal antibody against CD20+ B cells, has an overall response rate of 25-50%. It may be preferable to long-term steroid therapy. Rituximab eliminates normal B cells, including those that produce the antiplatelet antibody. The B cell depletion lasts for 12-18 months, and there have been few side effects or toxicities demonstrated.
Other agents that have induced responses include Rh0 (D) immune globulin, intravenous immune globulin, azathioprine, cyclophosphamide, danazol, vinca alkaloids (vincristine, vinblastine), dapsone, cyclosporine, and mycophenolate mofetil.37
For acute bleeding episodes, bleeding should be controlled with high-dose steroid therapy (methylprednisolone 1-2 grams IV/day) and with immune globulin at a dose of 1 gram per kilogram per day for two or three consecutive days.30 Phagocytic blockage may be accomplished with either intravenous immune globulin (IVIG) or anti-Rh0D immune globulin (25-75 µg/kg for two consecutive days). IVIG may contain anti-idiotypic antibodies that impede antibody production. It should be noted that IVIG can cause meningismus and headache, prompting consideration of lumbar puncture. Platelet transfusion should be withheld until the first dose of steroids has been given but may be used in emergencies to treat bleeding. An infusion of platelets should be two to three times the usual dose infused.30 The platelets associated with ITP are young and metabolically active as detected by flow cytometry, offering an explanation for the fact that bleeding is typically less pronounced with ITP than in states of bone marrow failure at similar platelet counts.38 Antifibrinolytic therapy with aminocaproic acid may reduce mucosal bleeding, and recombinant factor VII may be considered.39
The decision to treat a child with ITP is based upon the unproven assumption that shortening the duration of severe thrombocytopenia (platelet counts below 10,000-20,000/µL) affords protection from intracranial hemorrhage. The case for observation is that most children with typical ITP recover completely within a few weeks.30
Small pilot studies have investigated the use of thrombopoietic agents. A non-immunogenic thrombopoietic agent, AMG 531, administered subcutaneously for 3 to 6 weeks showed an overall response rate of 68%a rise in platelet count to greater than 50,000/µL, or at least twice the baseline countin a pilot study.40
TTP-HUS-DIC
Three related syndromes are important to diagnose in a patient with thrombocytopenia.
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening disease that typically presents with fever, neurologic abnormalities, and renal dysfunction in addition to thrombocytopenia. It occurs primarily in adults. Platelet microthrombi form throughout the body leading to clogging of the vasculature. Most vulnerable are the kidney, brain, heart, and adrenal glands. However other organ systems are affected. Untreated, the mortality rate is as high as 90%, although with aggressive treatment 80-90% survive.
A related syndrome, hemolytic uremic syndrome (HUS), occurs primarily in children with hemolytic anemia, thrombocytopenia, and acute renal failure. This disease often occurs after a viral illness or infectious diarrhea. Despite thrombocytopenia, bleeding is rare.
Finally, disseminated intravascular coagulation (DIC) presents as a systemic disease usually associated with sepsis or severe illness. Platelets are low and bleeding is common.
Common to all of these diseases is the finding of schistocytes on the peripheral blood smear along with decreased and large (young) platelets.
Heparin-Induced Thrombocytopenia
First described in 1958, heparin-induced thrombocytopenia (HIT) is a life-threatening disorder that follows exposure to unfractionated heparin (UFH) or, less commonly, low-molecular weight heparin (LMWH). Classically, patients present either with a low platelet count (less than 150,000/µL) or a relative decrease of 50% or more from baseline.41 Deep venous thrombosis and pulmonary embolism are its most frequent sequelae, although arterial eventsloss of limb, stroke, and myocardial infarctioncan also occur.42 Differences in the absolute platelet count greater than 70,000 to 90,000/µL infrequently occur within the same patient. Therefore, if a patient's platelet count has recently fallen 50% or more from a prior value, this should raise the strong possibility of HIT in any patient begun on heparin therapy within the preceding five to 10 days, especially with clinical evidence for new thrombosis. Resistance to UFH, defined as an inability to maintain a therapeutic activated PTT despite increasing dosage, may herald HIT.43
Heparin-induced thrombocytopenia is caused by antibodies against complexes of platelet factor 4 (PF4) and heparin. These antibodies also may be present in patients exposed to heparin but in whom no clinical manifestations develop. The time of onset of thrombocytopenia after initiation of heparin is typically 5-10 days in patients who have had no exposure to heparin or have had remote (over 100 days) exposure. Precipitous declines in platelet counts may occur if patients have had a recent exposure to heparin and have detectable levels of PF4-platelet antibodies.44 Platelet levels seldom drop below 10,000/µL, and typically recover within 4-10 days. It is not uncommon; the incidence of HIT may be as high as 5% in patients receiving UFH, and approximately 1% in patients receiving LMWH.41 It is important to note that heparin flushes, prophylactic doses of heparin subcutaneously, or small amounts of heparin on coated catheters, all can cause HIT.
Type I HIT is non-immune mediated and entails a small transient drop in platelet count between days 1 and 4 of treatment. Type II HIT is an immune-mediated process that can result in thromboembolic complications and death.42
HIT is seldom associated with bleeding. However, the thrombotic risk is more than 30 times that of control populations.45 The risk of thrombosis remains high for days to weeks after discontinuation of heparin, even after the platelet count normalizes.46 HIT may manifest as skin necrosis, venous gangrene of the limbs, and anaphylactic-type reactions after intravenous heparin. Thrombosis may develop one to seven days before an apparent fall in the platelet count. Thromboses may occur in either venous or arterial beds, with venous thromboembolism occurring four times as often as arterial events. Limb ischemia has resulted in amputation in 5-10% of patients with HIT. Venous thromboses predominate in medical and orthopedic patients, while arterial thromboses occur frequently in patients who have undergone cardiac or vascular surgery, including multiple saphenous grafts causing myocardial infarction.47 Thromboses may occur at unusual sites, such as in adrenal veins causing hemorrhagic necrosis of the adrenal gland, or in cerebral venous sinuses.48 Thrombotic risk appears to be greater among patients with higher levels of PF4-heparin antibody, or with a drop in platelet levels of more than 70%.49
The incidence of HIT appears to be approximately 10 times higher in patients receiving UFH, as opposed to LMWH. However, patients receiving LMWH have a more frequent incidence of HIT if they have had a recent exposure to UFH (within 100 days).50
The clinical diagnosis of HIT depends upon ruling out other causes for thrombocytopenia, such as infection, bone marrow disease, DIC, post-transfusion purpura, and drugs other than heparin. DIC may occur along with HIT, characterized by hypofibrinogenemia, prolonged INR and PTT, low levels of antithrombin and protein C, renal failure, and schistocytes on the peripheral smear. Platelet counts should recover after the discontinuation of heparin. Laboratory diagnosis includes testing for heparin-dependent antibodies with the use of serologic or functional assays.42
Serologic assays are available at most clinical laboratories; they detect circulating IgG, IgA, and IgM antibodies. Immunoassays have high sensitivity (more than 97%), but their specificity (74-86%) is limited by the fact that they also detect PF4-heparin antibodies in patients who do not have HIT. The negative predictive value of serologic assays is high.48,51
To confirm HIT in patients with positive serologic assays, functional assays measuring platelet activation to detect heparin-dependent antibodies capable of binding to and activating the Fc receptors on platelets may be performed. The serotonin release assay is considered the gold standard among the washed-platelet tests, but may not be readily available in all laboratories.52 The positive predictive value of functional assays tends to be high (89-100%).53 A rapid (within 30 minutes) and reliable HIT antigen assay may be available soon.42
Management of HIT centers on reduction of the thrombotic risk by reducing platelet activation and thrombin generation. All sources of heparin, including intravenous heparin used to keep lines open and LMWH, should be discontinued. Alternative anticoagulant therapy should be initiated and tailored to the patient's condition. Warfarin monotherapy is contra-indicated, due to reports of venous gangrene in the limbs, and warfarin-induced skin necrosis.54
The mainstay of treatment for HIT centers on anticoagulation with one of two classes of anticoagulants: direct thrombin inhibitors (DTI) or heparinoids.48 Direct thrombin inhibitors bind and inactivate thrombin. Three are currently available: lepirudin, argatroban, and bivalirudin. Lepirudin is a recombinant analogue of hirudin, a leech protein. It is metabolized by the kidney; the dose must be adjusted if the patient has renal insufficiency. Fatal anaphylaxis has been reported after sensitization to lepirudin, and patients should not be treated with this agent more than once. Argatroban is a small synthetic molecule that binds reversibly to the catalytic site of thrombin. As with lepirudin, the benefit to treatment appears to be mainly in the reduction of new thromboembolic complications, as opposed to reduction in death or amputation.55 Both lepirudin and argatroban can be monitored by the aPTT, which should be 1.5-3 times the baseline level for argatroban, and 1.5 to 2.5 times the baseline level for lepirudin. Argatroban is metabolized in the liver; dose adjustments are recommended in patients with moderate liver disease. It has been demonstrated to decrease the likelihood of new stroke and stroke-associated mortality in HIT.56 Only argatroban and lepirudin are FDA approved for the management of HIT.42
Bivalirudin is another synthetic thrombin inhibitor approved by the Food and Drug Administration for percutaneous coronary intervention in patients who have or are at risk for HIT. Its action is monitored by measuring the PTT or by following the activated clotting time (ACT). Danaparoid, a mixture of heparin sulfate and dermatan sulfate, another inhibitor of activated factor X, is available in Canada and Europe, but not in the United States.48
Therapy with an alternative anticoagulant agent should be followed by a transition to warfarin, but only after platelet counts have recovered to more than 150,000/µL. Oral anticoagulants should be overlapped with a direct thrombin inhibitor until the international normalized ratio (INR) is therapeutic for at least 48 hours.57 PF4-heparin antibodies disappear from the circulation within a median of 85 days.44 Documentation of heparin-induced thrombocytopenia should be included in the patient's medical record, and future exposure to heparin generally should be avoided. For certain procedures, such as cardiac bypass surgery, the use of direct thrombin inhibitors poses a considerable bleeding risk. Patients with a remote history of HIT with negative tests for PF4-heparin antibodies may therefore receive heparin during the procedure.48
Re-exposure to UFH may be safe after 100 days from the last heparin dose, by which time the UFH-dependent antibody will have disappearedprovided that exposure to UFH is brief, and that it can be demonstrated that no antibody is present.42
Platelet Transfusions
In the early 1980s and 1990s, use of platelet transfusion increased rapidly in the United States, doubling in the decade of the 1980s alone.58 This increased use of platelets correlates with increasingly aggressive myelosuppressive therapy for malignancies and increased availability of platelets because of cost-effective methods for storage of platelet concentrates. During the past 2 decades, technical improvements have doubled the number of platelets in each unit. The range around this average is high: 0.4-1.8 x 1011 related to the variability in donor platelet level.59,60 For this reason, at least 5 units must be pooled to ensure that the pool contains at least 3 x 1011 platelets. Platelet recovery and survival is satisfactory after 7 days of storage61; however, because of bacterial contamination, it has been recommended that the storage period be only 5 days.
It has been estimated that the average patient has an increase in platelet concentration of 10,000/µL per square meter of body surface area (BSA) per whole-blood-derived unit of platelets. Therefore, an adult with a BSA of 2 m2 should have a rise in platelet count of 5000/µL for each unit transfused. One unit is adequate only for transfusion of a small child weighing less than 14 kg. For transfusion of adults, 4 to 8 units must be pooled to provide a therapeutic dose. No standard dose fits all patients, regardless of size and clinical situation. For example, 4 units might be enough for prophylaxis in a patient with a BSA of 2 m2 who is not bleeding, to raise the platelet concentration from 5000/µL to the range of 20,000-30,000/µL.62 On the other hand, a person undergoing an invasive procedure or who is actively bleeding might be better served with a 10 unit transfusion to attain a platelet concentration of more than 50,000/µL.63
The traditional platelet concentration that should trigger a platelet transfusion has been 20,000/µL, but studies have shown that this level may be lowered to 10,000/µL in patients with production levels that are stable.64 Crossmatching is unnecessary for platelet transfusions. However, Rh negative patients should receive Rh negative platelets due to the potential for Rh sensitization. Each bag contains at least 5.5 x 10 platelets in 50-70 mL of plasma. The usual transfusion dose in adults is 6 to 10 units. In children it is 1 unit per 10 kg body weight. If human leukocyte antigen (HLA) matching of platelets is required, leukocyte-reduced apheresis platelets may be administered to prevent HLA antibody formation.62
Complications from platelet transfusion most frequently result from contaminating leukocytes, red cells, plasma proteins, and micro-organisms. (See Table 4.) The frequency of complications resulting from contaminating leukocytes can be reduced by prestorage leukoreduction of the platelet products. HLA alloimmunization can be reduced by consistent use of leukoreduced blood products.65,66
 |
Two specific situations are of particular relevance to the emergency physician. Massive trauma may necessitate large amounts of blood transfusions, causing dilutional thrombocytopenia. Even after one to two blood volumes are replaced, however, the platelet count is likely to be not less than 50,000/µL. Therefore, unless abnormal bleeding is noted, transfusion of platelets is generally unnecessary.67 Secondly, the patient with ITP generally does not require platelet transfusion because the bleeding tendency tends not to be severe relative to thrombocytopenic disorders resulting from decreased production. Furthermore, there is generally a satisfactory and rapid response to medical therapy. If critical bleeding occurs or surgery is required, 3 to 6 units of platelet concentrates per square meter of BSA generally raises the platelet count for 12 to 48 hours, recognizing that the survival of these transfused platelets is relatively brief.68
Resolution of Cases
Case 1. It is clear that this patient has isolated thrombocytopenia, without other evidence for bone marrow depression. Because the hospital had no in-patient beds, the patient stayed overnight in the Clinical Decision Unit (CDU) pending placement in a detoxification center. He is transfused with 10 units of platelets overnight. His repeat platelet count the next day is 55,000/µL. You are satisfied that his platelets are not undergoing immune destruction and that his thrombocytopenia is secondary to a combination of marrow suppression of platelet production by alcohol, congestive splenomegaly, and folate deficiency. After one week without alcohol intake in a detoxification unit, his platelet count is 113,000/µL. He never requires any further work-up.
Case 2. Although you are fairly certain this child has idiopathic thrombocytopenia due to the lack of any symptoms except for petechiae, he is admitted to the pediatric service for a diagnostic work-up. The child undergoes a bone marrow biopsy, as the consulting hematology service is worried about the possibility of leukemia. After the biopsy, he is started on high-dose steroids. The biopsy demonstrates no evidence for leukemia, and his platelet count improves after 4 weeks to 95,000/µL.
Case 3. Heparin-induced thrombocytopenia is not uncommon shortly after discharge from the hospital for a thrombotic event treated with UFH. You know not to simply increase the patient's warfarin, and start her instead on argatroban. While in the hospital, she does indeed have a Greenfield filter placed in her inferior vena cava, and is kept on warfarin for an additional 6 months. She recovers uneventfully from the pulmonary embolus.
Summary
When thrombocytopenia is discovered, the physician should attempt to determine the cause and assess the risk of bleeding. The most important laboratory test is the peripheral smear. Platelet counts of less than 100,000/µL are considered thrombocytopenic, and the bleeding time increases proportionately with a decrease in count below this level. Patients with platelet counts below 5000-10,000/µL are at risk for spontaneous bleeding, and should be admitted. Platelet counts above 50,000/µL are usually sufficient to control bleeding caused by local pathology or by trauma, provided that platelet function is normal.
Platelet transfusions should be initiated if there is active bleeding unresponsive to local measures and the platelet count is less than 50,000/µL. Prophylactic transfusion should be considered to prevent intracranial hemorrhage if the platelet count is below 10,000/µL. There are several reasons not to transfuse platelets unnecessarily: cost, disease transmission, and avoidance of allo-immunization, most prominentlyespecially if bone marrow transplantation in the future is a possibility. Immune destruction of platelets may occur in patients with certain disorders, but transfusion may be lifesaving in the actively bleeding patient. The mainstay of diagnosis in thrombocytopenia for the emergency physician is the history, exam, CBC, and peripheral smear.
Conclusions
Thrombocytopenia is a common hematologic disorder that may herald systemic disease, and may be caused by a variety of toxic, immunologic, infectious, and neoplastic disorders. Therapy depends upon the presentation and the underlying disorder. The emergency physician should recognize initial treatment and, with the growth of observation medicine, be prepared to manage bleeding complications, prescribe medications for stabilization of the underlying process, and be familiar with transfusion issues.
References
1. Solanki D, Blackburn B. Spurious leukocytosis and thrombocytopenia. A dual phenomenon caused by clumping of platelets in vitro. JAMA 1983;250:2514.
2. Landaw SA, George SA. Approach to the patient with thrombocytopenia. Uptodate 2007, available at www.uptodateonline.com/utd/content/topic.do?topicKey=platelet. Accessed 11/13/07.
3. Leslie SD, Toy PTCY. Laboratory hemostatic abnormalities in massively transfused patients given red blood cells and crystalloid. Am J Clin Pathol 1991;96:770.
4. Aster RH. Pooling of platelets in the spleen: Role in the pathogenesis of "hypersplenic" thrombocytopenia. J Clin Invest 1966;45:645.
5. Wang CS, Yao WJ, Wang ST, et al. Strong association of hepatitis C virus infection and thrombocytopenia: Implications from a survey of a community with hyperendemic HCV infection. Clin Infect Dis 2004;39:790-796.
6. Jadavji T, Scheifele D, Halperin S. Thrombocytopenia after immunization of Canadian children, 1992 to 2001. Canadian Paediatric Society/Health Canada Immunization Monitoring Program. Pediatr Infect Dis J 2003;2292:119-122.
7. Ladhani S, Khatri P, El-Bashir H, et al. Imported malaria is a major cause of thrombocytopenia in children presenting to the emergency department in east London. Br J Haematol 2005;129:707.
8. Chintagumpala M. Approach to the child with thrombocytopenia. UpToDate, available at www.uptodateonline.com/utd/content/topicKey=pedi_hem/9749&view=text. Accessed 11/13/2007.
9. Ballmeier M, Germeshausen M, Chulze H, et al. C-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001;97:139.
10. Hedberg VA, Lipton JM. Thrombocytopenia with absent radii. A review of 1000 cases. Am J Pediatr Hematol Oncol 1988;10:51.
11. Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype. Blood 1997;90:2680.
12. Lacey JV, Penner JA. Management of idiopathic thrombocytopenic purpura in the adult. Semin Thromb Hemost 1977;3:160.
13. Howard SC, Gajjar A, Ribeiro RC, et al. Safety of lumbar puncture in children with acute lymphoblastic leukemia and thrombocytopenia. JAMA 2000;284:2222-2224.
14. Reid DM, Shulman NR. Drug porpora due to surreptitious quinidine intake. Ann Intern Med 1988;108:206-208.
15. Vanderschueren S, De Weerdt A, Malbrain M, et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med 2000;28:1871.
16. Drachman JG. Inherited thrombocytopenia: When a low platelet count does not mean ITP. Blood 2004;103:390.
17. George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: A practice guideline developed by explicit methods for the American Society of Hematology. Blood 1996;88:3-40.
18. Esparza SD, Moore TB, Feig SA. ITP and empiric steroid treatment. J Pediatr Hematol Oncol 2003;25:674.
19. Majhail NS, Lichtin AE. What is the best way to determine if thrombocytopenia in a patient on multiple medications is drug-induced? Cleve Clin J Med 2002;69:259-262.
20. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886-890.
21. Pedersen-Bjergaard U, Andersen M, Hansen PB. Thrombocytopenia induced by noncytotoxic drugs in Denmark 1968-91. J Intern Med 1996;239:509.
22. Aster R. Drug-induced thrombocytopenia. In: Michelson AD, ed. Platelets. New York: Academic Press 2007:887-902.
23. Bougie D, Aster R. Immune thrombocytopenia resulting from sensitivity to metabolites of naproxen and acetaminophen. Blood 2001;97:3846-3850.
24. Aster RA, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580-587.
25. Berkowitz SD, Harrington RA, Rund MM, et al. Acute profound thrombocytopenia after c7E3 Fab (abciximab) therapy. Circulation 1997;95:809.
26. Pedersen-Bjergaard U, Anderson M, Hansen PB. Drug-specific characteristics of thrombocytopenia caused by non-cytotoxic drugs. Eur J Clin Pharmacol 1998;54:701.
27. George JN. Drug-induced thrombocytopenia. UpToDate, available at http://www.uptodate.com/utd/content/topic.do?topicKey=platelet/4585&view=text. Accessed 11/13/2007.
28. Neylon, Saunders PW, Howard MR, et al. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: A prospective study of a population-based cohort of 245 patients. Br J Haematol 2003;122:966.
29. Azuno Y, Yaga K, Sasayama T, et al. Thrombocytopenia induced by Jui, a traditional Chinese herbal medicine. Lancet 1999;354:304-305.
30. Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002;346:995-1008.
31. Bromberg ME. Immune thrombocytopenic purpurathe changing therapeutic landscape. N Engl J Med 2006;355:1643-1645.
32. Nieminen U, Peltola H, Syrjala MT, et al. Acute thrombocytopenic purpura following measles, mumps and rubella vaccination. A report on 23 patients. ACTA Paediatr 1993;82:267.
33. Chintagumpala M. Approach to the child with thrombocytopenia. 2007 UpToDate, available at http://www.uptodateonline.com/utd/content/topic.do?topicKey=pedi_hem/9749&view=text. Accessed 11/13/2007.
34. Lilleyman JS. Intracranial haemorrhage in idiopathic thrombocytopenic purpura. Paediatric Haematology Forum of the British Society for Haematology. Arch Dis Child 1994;71:251.
35. Brighton TA, Evans S, Castaldi PA, et al. Prospective evaluation of the clinical usefulness of an antigen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood 1996;88:194-201.
36. Warner MN, Moore JC, Warkentin TE, et al. A prospective study of protein-specific assays used to investigate idiopathic thrombocytopenic purpura. Br J Haematol 1999;104:442-447.
37. Vesely SK, Perdue JJ, Rizvi MA, et al. Management of adult patients with persistent idiopathic thrombocytopenic purpura following splenectomy: A systematic review. Ann Intern Med 2004;140:112-120.
38. Saxon BR, Mody M, Blanchette VS, et al. Reticulated platelet counts in the assessment of thrombocytopenic disorders. Acta Paediatr Suppl 1998;424:65-70.
39. Vidarsson B, Onundarson PT. Recombinant factor VIIa for bleeding in refractory thrombocytopenia. Thromb Haemost 2000;83:634-635.
40. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672-1681.
41. Warkentin TE, Roberts RS, Hirsh J, et al. An improved definition of immune heparin-induced thrombocytopenia in postoperative orthopedic patients. Arch Intern Med 2003;163:2518-2524.
42. Bartholomew JR, Begelman SM, Almahameed A. Heparin-induced thrombocytopenia: Principles for early recognition and management. Cleve Clin J Med 2005;72:S31-S36.
43. Buckley MF, James JW, Brown DE, et al. A novel approach to the assessment of variations in the human platelet count. Thromb Haemost 2000;83:480.
44. Warkentin TE, Kelton JG. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med 2001;344:1286-1292.
45. Girolami B, Prandoni P, Stefani PM, et al. The incidence of heparin-induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: A prospective cohort study. Blood 2003;101:2955-2959.
46. Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med 1996;101:502-507.
47. Ayala E, Rosado MF, Morgansztern D et al. Heparin-induced thrombocytopenia presenting with thrombosis of multiple saphenous vein grafts and myocardial infarction. Am J Hematol 2004;76:383-385.
48. Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. N Engl J Med 2006;355:809-817.
49. Greinacher A, Farner B, Kroll H, et al. Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis: A retrospective analysis of 408 patients. Thromb Haemost 2005;94:132-135.
50. Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: A meta-analysis. Blood 2005;106:2710-2715.
51. Warkentin TE, Sheppard JA, Moore JC, et al. Laboratory testing for the antibodies that cause heparin-induced thrombocytopenia: How much class do we need? J Lab Clin Med 2005;146:341-346.
52. Chong BH. Heparin-induced thrombocytopenia. J Thromb Haemost 2003;1:1471-1478.
53. Pouplard C, Amiral J, Borg JY, et al. Decision analysis for use of platelet aggregation test, carbon 14-serotonin release assay, and heparin-platelet factor 4 enzyme-linked immunosorbent assay for diagnosis of heparin-induced thrombocytopenia. Am J Clin Pathol 1999;111:700-706.
54. Warkentin TE, Elavathil LJ, Hayward CP, et al. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Intern Med 1997;127:804-812.
55. Lewis BE, Wallis DE, Leya F, et al. Argatroban anticoagulation in patients with heparin-induced thrombocytopenia. Arch Intern Med 2003;163:1849-1856.
56. LaMonte MP, Brown PM, Hursting MJ. Stroke in patients with heparin-induced thrombocytopenia and the effect of argatroban therapy. Crit Care Med 2004;32:976-980.
57. Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia: Recognition, treatment, and prevention. The seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004:126 Suppl:311S-337S.
58. Surgenor DM, Wallace EL, Hao SHS, et al. Collection and transfusion of blood in the United States, 1982-1988. N Engl J Med 1990;322:1646.
59. Kelley DL, Fegan RL, Ng AT, et al. High yield platelet concentrations attainable by continuous quality improvement reduce platelet cost and donor exposure. Transfusion 1997;37:482.
60. Hoeltge GA, Shah A, Miller JP. An optimized strategy for choosing the number of platelet concentrates to pool. Arch Pathol Lab Med 1999;123:928.
61. Rock G, Neurath D, Cober N, et al. Seven-day storage of random donor concentrates PLT. Transfusion 2003;43:1374.
62. Vassallo R, Murphy S. Preservation and clinical use of platelets, Chapter 132. In: Lichtman MA, Kipps TJ, Kaushansky, eds. Williams Hematology, 7th ed. McGraw-Hill: New York; 2006: 2175-2188.
63. McVay PA, Toy PT. Lack of increased bleeding after liver biopsy in patients with mild hemostatic abnormalities. Am J Clin Pathol 1990;94:747.
64. Contreras M. The appropriate use of platelets: An update from the Edinburgh Consensus Conference. Br J Haematol 1998;101:10.
65. [No authors listed.] Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. The Trial to Reduce Alloimmunization to Platelets Study Group. N Engl J Med 1997;337:1861.
66. Seftel MD, Growe GH, Petraszko T, et al. Universal prestorage leukoreduction in Canada decreases platelet alloimmunization and refractoriness. Blood 2004;101:333.
67. Reed RL, Ciavarella D, Heimbach DM, et al. Prophylactic platelet administration during massive transfusion. A prospective, randomized double-blind clinical study. Ann Surg 1986;203:40.
68. Carr JM, Kruskall MS, Kaye JA, et al. Efficacy of platelet transfusions in immune thrombocytopenia. Am J Med 1986;80:1051.
Most cases of thrombocytopenia seen in the emergency department (ED) are expected. Patients are known to have hematological disease or are receiving chemotherapy. At times, however, the ED physician is confronted with an unexpected laboratory finding in an assymptomatic patient, or with a patient who is bleeding. The challenge, as usual, is to determine the need for acute treatment and the appropriate disposition.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.