Immigrant Medicine: The Emergency Department Perspective, Part II - Commonly Encountered Diseases of Latin America, Asia, and Africa
Part II: Commonly Encountered Diseases of Latin America, Asia, and Africa
Authors: Mary C. Meyer, MD, Attending Physician, Emergency Department, Kaiser Oakland and Kaiser Walnut Creek, Oakland, CA; Danica N. Barron, MD, Alameda County Medical Center, Highland General Hospital, Oakland, CA; R. Carter Clements, MD, FACEP, Assistant Chair, Department of Emergency Medicine, Alameda County Medical Center—Highland Campus, Oakland, CA; Clinical Instructor, Department of Internal Medicine, University of California, San Francisco.
Peer Reviewers: Judith C. Brillman, MD, FACEP, Associate Professor, Research Co-Director, Department of Emergency Medicine, University of New Mexico School of Medicine, Albuquerque; Hans House, MD, DTMU, Assistant Professor, Program in Emergency Medicine, University of Iowa, Iowa City.
President John F. Kennedy once wrote that the United States is a "nation of immigrants."1,2 Ninety-nine percent of the current U.S. population has descended from prior immigrants,3 and foreign-born individuals now represent 11% of the U.S. population.4 In the 1990 United States census, the immigrant population totaled 19.8 million, an all-time high.5 It is estimated that an additional 7 million undocumented aliens currently are residing in the United States.4 In 1993 alone, 300,000 illegal residents gave birth to children who became U.S. citizens.6
The demographics of our foreign-born population have undergone a polar shift during the past 50 years. Europeans once made up the majority of new arrivals, but Asians and Latin Americans now predominate.5,6 In 1988, 84% of documented new arrivals were from either Asia or Latin America.7 Recent data indicate that the top five countries sending legal immigrants to the United States are Mexico, the Philippines, Vietnam, China, and Korea.6 These figures do not reflect the substantial population of undocumented aliens residing in the United States, most originating from Latin America.6 Africa and the Middle East are increasing in overall proportion as sources of new immigrants to the United States. During the past two decades, there have been almost 10,000 new arrivals from Ethiopia alone.8 Finally, the majority of the 8000 international children adopted annually were born in the developing world.8
Foreign-born patients often bring with them a host of "exotic" diseases specific to their countries of origin. Unlike Western patients who suffer from industrial diseases such as cancer, stroke, and coronary artery disease, the predominant diseases of immigrants traditionally arise from infections and parasites, malnutrition, and lack of access to basic health care.6,8 This is changing somewhat as globalization brings high-fat diets and tobacco use to developing nations. Proper care of these patients requires a familiarity with their unique disease patterns and an understanding of variations in local immigrant demographics. New York City and Los Angeles, for instance, have significant populations of Soviet refugees, whereas San Francisco claims one of the largest Vietnamese populations outside Vietnam.
Emergency physicians (EPs) in the United States receive scant formal training about diseases of the tropical world.8 Working with immigrant patients is challenging due to the protean manifestations of the diseases of the developing world and is exacerbated by basic cultural and language barriers. In Part I of this series, common diseases found throughout the developing world were discussed and reviewed. Part II of this series will familiarize physicians with some of the infections and diseases specific to immigrants from particular regions of the world. Often, a disease process in an immigrant patient may be predicted by nationality of origin; for instance, Brazilian children with ascites are assumed to have hepatosplenic schistosomiasis, and in Mexico, adults with new onset seizures are screened for neurocysticercosis.8 It is important to remember that while certain illnesses may predominate in some geographic regions, they may occur sporadically around the globe. An exhaustive review of exotic diseases is beyond the scope of this article. Therefore, the emphasis of this article is on the most frequently encountered maladies and the most classic presentations of each disease. Tropical infections are, by nature, notorious for their varied and unusual manifestations.8 For the purpose of simplicity, this article groups foreign-born individuals by region of origin: Latin America; Asia and South Asia; or Africa. Table 1 lists the commonly encountered exotic diseases by major areas of distribution, and Table 2 lists some of the tropical etiologies of various patient complaints.—The Editor


Latin America
Neurocysticercosis. Epidemiology. Cysticercosis is a parasitic disease caused by infection with the larval stage of the pork tapeworm Taenia solium.9,10 In humans, virtually every organ system may be affected. Larvae migrate into tissues and then produce cysts that later become calcified nodules. Encystment of subcutaneous tissues and muscular tissues is most common, but generally is asymptomatic. More clinically relevant is infection of the central nervous system, producing the disorder known as neurocysticercosis (NCC). NCC is the most common parasitic infection of the central nervous system. While it does occur in Africa and Asia, the disease is particularly prevalent in countries of Latin America.9 It has been estimated that 2.5-3.6% of the population of Mexico is infected with T. solium.9-12 In Mexico, NCC is the leading cause of all neurological admissions to hospitals, accounting for 10% of these admissions.9 It also is the leading cause of adult-onset seizures in Latin America, where the incidence of epilepsy is approximately twice that of developed nations.9,13
In the United States, rates of T. solium infection have been on the rise for several decades, a phenomenon attributed to steadily rising numbers of immigrants from Latin America. Studies suggest that the vast majority of NCC encountered in the United States occurs in the Latino foreign-born population.11 One review found that of 500 NCC cases diagnosed in four Los Angeles area hospitals, 95% occurred in Latinos.9 Multiple other studies from the southwest United States have found that approximately 90% of NCC cases occurred in immigrants from Mexico or South America.9
To date, the greatest number of documented NCC infections is to be found in the southwestern United States, with California leading the way.8 There are, of course, occasional exceptions. In 1995, a community of Orthodox Jews in Brooklyn experienced an outbreak of T. solium infection.8,14,15 Epidemiologists were baffled at first, since the Orthodox Jewish community prohibits consumption of pork. The outbreak ultimately was traced to domestic workers from Mexico and Central America who harbored intestinal tapeworm and had unknowingly contaminated the food and water supplies of their Jewish employers.8,14-16 In the emergency department (ED), it is important to maintain a high level of suspicion for NCC in any Latino immigrant who presents with new onset seizures or other neurological complaints.
Pathology. Infection with T. solium may result in either intestinal tapeworm disease or cysticercosis, depending on the route of infection. Cysticercosis spreads via person-to-person contact as a result of fecal contamination of food or water supplies.10,11,17 The cycle begins when an individual harboring intestinal tapeworm sheds T. solium eggs into his stool.10,11,17 If the eggs are ingested by a person or pig, they will hatch into larvae in the lumen of the intestine. The larvae then penetrate the bowel wall and disseminate to various organs, most often the brain, muscles, and subcutaneous tissue.10,11 Occasionally, retinal tissue or the spinal cord also will be involved.9,10,16 Once inside human (or pig) organs, the larvae encyst and the result is multiple cysticerci, or fluid-filled vesicles containing viable larvae.10,17 Alternately, human beings may develop intestinal tapeworm by consuming undercooked pork meat infected with T. solium.11 Ingested cysticerci from the pork meat decyst in a human’s intestine and attach to the wall of the bowel, maturing into a tapeworm that ultimately releases eggs into the person’s stool.10,11
The term "neurocysticercosis" refers specifically to cysticerci found within the central nervous system.9,11 Within the brain, cysts may be solitary or multiple and may occur in many different areas. The most common sites of cysticerci infection include the cerebral cortex, basal ganglia, subarachnoid space, and ventricles.9,10 As a rule, the fibrous outer layer of the cyst initially prevents a significant immunologic response; therefore, the cysticeri can exist unharmed for years in human tissue.10,11 Some patients will tolerate hundred of cysts with minimal symptoms. After a time, usually 2-5 years, the cysticerci undergo spontaneous degeneration and calcify.9,16-18 It is during this degeneration process that the cysts first provoke an inflammatory response from the human host; lymphocytes and eosinophils migrate to surrounding tissue, causing significant local edema.9,19 NCC has a wide spectrum of clinical manifestations, varying with the tissue burden of cysts and the host response.16 Symptoms may occur during all stages of infection, secondary to mass effect of the cysticerci or calcifications in brain tissue. As degenerating cysts provoke a ring of surrounding edema, they tend to be associated with more severe effects.
Clinical Spectrum. The most common presenting symptom of NCC infection is epilepsy.20 Seizures, which may be generalized or focal, occur in 50-80% of affected individuals and commonly are the only manifestation.9-11,18 The neurological exam typically is normal. Nonetheless, signs and symptoms of the disease may include headache, motor or sensory deficits, cerebellar ataxia, and involuntary movements.16,21 Symptoms typically will be subacute or chronic, and may wax and wane over time, correlating with the natural history of the T. solium cysts. Occasionally, subarachnoid cysts will produce enough inflammation to occlude the cerebral arteries, causing either a lacunar or large artery stroke.16,20 In Latin America, this complication is common, and NCC is a risk factor for stroke in the young.10 Ventricular cysts may result in hydrocephalus and increased intracranial pressure, with mental status changes, focal signs, nausea/vomiting, or intense intractable headaches.16 Occasionally, massive infection results in cysticercotic encephalitis; this disease, similar in presentation to other encephalitides, typically occurs in children and adolescents.9,16 There even are case reports of psychiatric disorders that ultimately were attributed to infection with T. solium.16 Spinal cord cysts occur in 1-5% of patients with cysticercosis, resulting in radiculopathy, paresthesias, and motor deficits at various cord levels.9,16
Diagnosis. Diagnosis of the disease usually entails a combination of imaging studies and serologic tests. Definitive diagnosis requires brain biopsy with identification of the parasite, but this step almost never is indicated with modern imaging techniques.11,16,17,21 Typically, the combination of imaging studies and serology is considered specific enough to initiate treatment.10,11
The initial modality of choice is a computerized tomography (CT) scan of the brain. Viable cysticerci appear on a CT scan as round cystic lesions that do not enhance with contrast, whereas degenerating larvae are visible as focal enhancing lesions with surrounding edema.9 Forty-four percent of cysts will demonstrate a visible scolex, or "head" of the parasite, inside.9 Dead parasites are seen as scattered calcifications within the brain parenchyma.9 The most characteristic findings of NCC are multiple cystic lesions and calcifications at various stages in the parasite’s life cycle;21 the presence of one or several ring-enhancing lesions should prompt a work-up for other causes, such as tuberculosis, malignancy, abscess, or toxoplasmosis.10,11,22 CT scan of the brain also may demonstrate changes consistent with hydrocephalus or infarct.9
Magnetic resonance imaging (MRI) is more sensitive than the CT scan at identifying live forms of the parasite,23,24 although it is less sensitive for detection of the presence of calcifications. It is particularly useful in delineating brainstem cysts, intraventricular cysts, and spinal cord lesions.10,23,24 One approach to neuroimaging suggests the use of CT of the brain as a screening tool. If the CT is negative and the presentation is clinically concerning for NCC, an MRI may be performed.9,16
If neuroimaging suggests infection with T. solium, serologic testing for anticysticercal antibodies can help confirm the diagnosis. Multiple assays now are available; the most effective is the immunoblot, with a sensitivity of 100% and a specificity of 98%.25,26 Sensitivity, however, falls significantly (40-65%) in patients with a single cyst or calcifications only; in these individuals the immunoblot frequently is negative.22,27 Therefore, serologic tests are helpful when they are positive, but a negative assay does not rule out NCC.17 Lumbar puncture does not play a significant role in the work-up of NCC. Its main utility rests in its ability to exclude other diagnoses. Spinal fluid in a patient with NCC most commonly is normal, although a mild lymphocytic pleocytosis may occur. Also, patients with NCC almost never manifest fever or meningeal signs, except in the rare case of cysticercosis encephalitis.16 Stool studies may be helpful in patients with particularly heavy NCC infections. Interestingly, patients with NCC usually are not co-infected with the adult intestinal tapeworm, but there is some evidence that patients with massive infection also may have intestinal infection, with autoinfection contributing to the neurocysticercosis in these patients.25
Treatment. Treatment of NCC requires a cocktail of antiparasitic therapy, steroids, and symptom-controlling drugs. Antiparasitic therapy has been recommended by some experts for any patients with cysts on neuroimaging; patients with only calcifications are considered to have dead parasites and do not need drug treatment.16 However, this is controversial. Data from several studies have indicated that most cystic lesions, if given time, usually show spontaneous improvement, either healing with calcification or disappearing entirely.28 Furthermore, the effects of antiparasitic therapy on patient symptomatology are unclear.9,10,16 Some studies have suggested an improvement in seizures and involuntary movements following treatment, likely from decreased mass effect on the brain.29,30 Other series have failed to show a substantial improvement after therapy, particularly in patients with epilepsy.9,31 Consequently, several experts have opposed the routine use of antiparasitic therapy, arguing that it is unnecessary and may even worsen clinical outcomes by provoking an acute inflammatory response.31-33 For the EP, the decision to use an antiparasitic agent may be appropriate in certain patients but should not be taken lightly, and should be done in consultation with an infectious diseases expert. As a general rule, patients who are to be treated with antiparasitics require either admission to the hospital or close follow-up, as a small proportion of treated individuals can be expected to deteriorate during the first several days of treatment. Both praziquantel and albendazole are approved for treatment of the disease;34,35 several trials have demonstrated a slightly improved cure rate with albendazole.9,17,36 The recommended dosages are: praziquantel 50 mg/kg/day for 15 days or albendazole 15 mg/kg/day for 8 days.9,16,17,37 A course of albendazole has been shown to eradicate 75-90% of cysts on follow-up CT of the brain.9
Various experts have recommended that steroids be pre-administered to any patient receiving antiparasitic therapy.38 A high frequency of adverse reactions has been noted in patients who receive praziquantel or albendazole, including headache, vomiting, seizures, and occasionally, mental status changes.11,17 It has been postulated that these side effects are a consequence of the brain’s inflammatory response to dying parasites.10,11,17 Because steroids appear to decrease the incidence of adverse reactions, their co-administration has become standard of care, although there are no controlled trials to support this practice.10,38 Some physicians administer corticosteroids (dexamethasone 6-12 mg/day) several days prior to initiation of antiparasite therapy, whereas others start steroids only if undesirable symptoms occur during treatment.16
Steroids also are the mainstay of treatment for patients with cysticercotic encephalitis. In this group of patients, antiparasitics are contraindicated. It is believed that acute edema from dying parasites may cause herniation and, in fact, a number of deaths have occurred in children following anti-parasitic therapy.10,39 In patients with large subarachnoid cysts, the traditional therapy has been surgical removal with or without a ventriculoperitoneal shunt; however, recent evidence suggests that antiparasitic therapy alone may be effective.40 Nonetheless, antiparasitics should be administered with caution in this subgroup of patients, in whom inflammation can induce infarction.10
Finally, patients with seizures will require anti-epileptic therapy. Seizures usually are controlled easily with either phenytoin or carbamazepine.16,41 Many patients can be weaned off their anti-epileptics following successful eradication of brain cysts.10
Overall prognosis is good; with current therapy, the five-year mortality is approximately 10%.41
Amoebiasis. Epidemiology. Amoebiasis is a parasitic disease caused by the protozoa Entamoeba histolytica.42 Humans are the main hosts for the parasite, with infection occurring via fecal contamination of food and water supplies. Amoebiasis is endemic throughout Mexico, Central and South America, India, and Africa, and is the most common protozoan cause of death after malaria. Approximately 40-50 million cases of amoebic colitis and liver abscess occur annually, and 40,000-110,000 people die from their infection.42 It is estimated that 10% of the world’s population is infected by E. histolytica.43
In the United States, amoebiasis is predominantly an imported disease from Mexico and Latin America. At a county hospital in Los Angeles, 76% of reported cases occurred in Latin Americans.44 Similarly, a retrospective review at a Texas hospital revealed that 43 out of 49 patients diagnosed with amoebiasis were Hispanic; the most common country of origin was Mexico, followed by El Salvador, Honduras, and Venezuela.45 Of note, a high prevalence of gastronintestinal (GI) colonization with Entamoeba has been documented in male homosexuals in the United States.42 More recent data, however, indicate that the infecting organism in these individuals usually is Entamoeba dispar, a nonpathogenic strain.42,46 Thus, while male homosexuals can contract amoebiasis, particularly if they are co-infected with HIV, the disease still remains much more prevalent in the immigrant population.42
For reasons that are not entirely clear, amoebic liver abscess classically occurs in young men between 20 and 40 years of age.42 Indeed, the Zulu word for the disease is "isigwebedhla," or "disease of the strong, young men."45 The pathogenesis of amoebic liver abscess is not well known, but it appears that an altered immune response to E. histolytica may play a role. Alcohol consumption, in particular, appears to be an important immunosuppressive factor.47 Eighty percent of patients diagnosed with amebic liver abscess are men,42,44,45,48 and 25- 40% of patients report heavy alcohol use, defined as consumption of at least 150 g/d.47,48 At least two-thirds of patients relate a history of travel to an endemic area.47
Pathology. Infection with E. histolytica begins when humans ingest parasite cysts from food or water that has been contaminated by a human carrier.44 In the human GI tract, these cysts hatch and the emerging trophozoites attach to the mucosa of the colon44,48 where they cause colonic amoebiasis, inducing lesions ranging from nonspecific mucosal thickening to the classic flask-shaped amoebic ulcer expanding transversely into the submucosa.49 The cecum is the most common site of intestinal infection, followed by the ascending colon, rectum, sigmoid, and appendix.49 Alternately, some parasites may ascend the portal venous system toward the liver. Obstruction of portal vessels causes local hepatic necrosis and liquefaction, forming multiple small abscesses that eventually coalesce into one large liver abscess.44,48,49 In the vast majority of individuals, infection with amoebiasis is transient and asymptomatic.47-49 A small percentage of patients remain asymptomatic and become carriers who continue to secrete cysts into their stools, thus promoting transmission to other individuals.47,49 An even smaller percentage, in the range of 7-8%, become symptomatic with the infection; these individuals develop either amebic colitis or a hepatic abscess.45,49 Although amoeba occasionally will metastasize to more distant organs of the body, hepatic abscess is by far the most common extra-intestinal manifestation of infection.47,49
Clinical Spectrum. Intestinal infection with E. histolytica is typically either asymptomatic or the patient may complain of abdominal colic or change in bowel habits. Some patients, however, will develop fulminant rectocolitis, characterized by fever, weight loss, abdominal tenderness, and bloody mucoid stools. Intestinal amoebiasis also may cause complications such as hemorrhage, peritonitis, and perforation. The infection may cause fistulas involving the perineum, genitalia, and abdominal skin. Finally, inappropriate administration of corticosteroids may induce toxic megacolon, a rare but ominous complication.49
Ten percent of patients with clinical amoebiasis will develop amoebic liver abscess. As the presenting signs and symptoms of amoebic liver abscess may be vague and nonspecific, it is important for the EP to be aware of the clinical signs indicating high risk for the disease.45 Most patients will describe fever (77-85%) and abdominal pain (74-92%), which may or may not be localized to the right upper quadrant.42,44-46 GI symptoms occur in 10-35% and include vomiting, abdominal cramps, abdominal distention, diarrhea, or constipation.42,44,45,48 The patient may relate a history of right pleuritic chest pain or right shoulder pain secondary to diaphragmatic irritation. Weight loss and weakness also have been reported.42,45 Of note, a history of dysentery or bloody stools is rare. Most patients no longer have gastrointestinal colonization with Entamoeba at the time of hepatic abscess presentation.44 The most common physical findings are abdominal tenderness and hepatomegaly.48 The physician may note decreased breath sounds at the right lung base or a pleural rub.48 Jaundice is rare.48 Laboratory studies may be misleading as they may not show significant alterations of liver function tests.44,46,50 Alkaline phosphatase or transaminase levels may be mildly increased,44,50 but large elevations should prompt concern about pyogenic abscess.48 Most patients demonstrate a leukocytosis with white blood cell (WBC) counts of 15,000-17,000, but eosinophilia almost never occurs.44,45,48 Chest x-ray frequently is abnormal and may be misleading; amoebic liver abscess often is misdiagnosed as pneumonia on initial presentation.44,50 Typical radiograph findings include an elevated right hemidiaphragm or a right pleural effusion.42,50
Diagnosis. The main diagnostic dilemma in patients with E. histolytica is distinguishing between parasitic vs. pyogenic liver abscess.44,46 Historical factors may be helpful; patients with pyogenic abscesses typically are older (50-70 years) and often have a history of abdominal surgery or gallbladder disease.44,46 Imaging studies may demonstrate the hepatic mass but cannot differentiate between parasitic or bacterial disease. Either ultrasound or abdominal CT is acceptable as the initial imaging modality of choice,45,46 as each has an accuracy of greater than 90%.51,52 MRI seldom is indicated.48 A technetium scan may differentiate between pyogenic and amebic abscesses; pyogenic lesions appear as "hot" spots whereas amebic lesions are "cold" spots with a hot ring around them.48,51,53
A variety of serologic assays are available and can add useful information to the clinical picture. These serologic tests all measure the presence of antibody to E. histolytica.48,49 The assays, unfortunately, remain positive for years after treatment, and thus, cannot distinguish acute from past disease.46,48,54 Further, many individuals from endemic areas already have antibodies to Entamoeba, particularly if the prevalence of disease in their native country is high.42,48 Research now is focused on the development of a serum assay that measures Entamoeba antigen directly.42,48,54,55 Until then, treatment often proceeds based on presumption of parasitic over pyogenic disease due to the clinical presentation.
Treatment. The mainstay therapy for amoebic liver disease is medical management.45 Treatment with oral metronidazole (750 mg 3 times per day for 10 days) results in a 90% cure rate.44,45,48 The patient with amoebic liver abscess typically will show clinical improvement within three days, reflected by defervescence and reduced abdominal pain.42,45 Alternately, if the patient cannot take metronidazole or does not respond to initial therapy, oral chloroquine can be used.48 It is dosed at 600 mg once daily for two days, followed by 300 mg once daily for another 2-3 weeks.48,50 Following treatment with metronidazole or chloroquine, a luminal agent is administered.42,48,50 The luminal agent is believed to prevent a 10% relapse rate based on the theory that a small percentage of patients have GI colonization with Entamoeba and may reseed the liver.48 The most commonly used luminal agents are iodoquinol, paromomycin, and diloxanide furoate.49,50
Unlike therapy for pyogenic liver abscess, routine aspiration of amoebic liver abscess is not indicated.45 Patients who are considered high risk for complications, however, should receive either needle aspiration or percutaneous abscess drainage. Studies have not demonstrated superiority of one method over the other.48,56 Candidates for aspiration include those with abscess cavities greater than 5 cm and left hepatic lobe lesions, both of which have a high rupture rate.44,47,50 Patients who fail medical management may require drainage.42,48,57 Although the patient can be expected to show rapid clinical improvement, both CT scan and ultrasound are much slower to normalize. Imaging studies normalize over a course of 3-9 months; hence, follow-up studies may be needed.44,45,48
Untreated, amoebic liver abscess has a mortality of 82%.47 Factors associated with poor prognosis are listed in Table 3. Death results from rupture of the abscess and its associated complications.44 The most common site of rupture is into the pleural cavity, causing empyema and adult respiratory distress syndrome (ARDS) with sepsis.44 Rupture of the abscess into the peritoneum may cause peritonitis, ileus, and bacterial superinfection.44 Occasionally a hepatic abscess will rupture into the pericardium. Mortality in this event approaches 60%, usually from cardiac tamponade.44 Of note, one study demonstrated that 20% of amebic liver abscesses were ruptured at the time of presentation; thus, the EP must be vigilant in evaluating vague complaints in these high-risk individuals.44

Chagas’ Disease. Epidemiology. Chagas’ disease is caused by the protozoan flagellate, Trypanosoma cruzi, one of 20 species in the genus Trypanosoma.58 African sleeping sickness is caused by the morphologically similar Trypanosoma brucei subspecies, including T. brucei gambiense and T. brucei rhodesiense. The endemic areas of Chagas’ disease and African sleeping sickness do not overlap, and the two protozoan species have little in common in terms of transmission, pathogenesis, and the clinical courses of the diseases.
Chagas’ disease is endemic to Latin America, predominantly in Mexico and Central American countries. The insect vector tends to reside in cracks and holes of mud, wood, and stone houses; therefore, human trypanosomiasis primarily affects poor persons living in rural areas. Eighty-five percent of cases occur in children younger than 10 years of age, with a mean age of 4 years.59 The World Health Organization (WHO) estimates that 16-18 million people are infected with T. cruzi, and 45,000 persons die annually from Chagas’ disease.60
Chagas’ disease may have two phases: acute and chronic. The acute phase is most common in children and carries a case fatality rate of approximately 12%. Chronic disease occurs years after initial infection and is marked by myocardial involvement and esophageal and colonic dilatation. Overall, 10-30% of individuals with chronic T. cruzi infestation develop symptomatic Chagas’ disease. For reasons unknown, there is significant geographic variability in the incidence of symptomatic disease. Areas in the northern endemic range, including Venezuela, Colombia, Mexico, and countries of Central America, have lower rates of cardiac disease, megaesophagus, and megacolon than countries in the southern range (Argentina, Bolivia, Brazil, Chile, Paraguay, and Uruguay).58 The presence of T. cruzi-infected insect vectors has been documented in parts of the southern and western United States. Nevertheless, acute Chagas’ disease is rare in the United States, likely due to high housing standards and low vector density.58 On the other hand, recent years have seen a marked increase in the number of people in the United States with chronic T. cruzi infection. More than 6 million persons who immigrated to the United States between 1971 and 1996 arrived from endemic areas. Extrapolation of data from Latino blood donors and United States census data suggests that chronic Chagas’ disease affects at least 50,000-100,000 immigrants.58
Pathology. The life cycle of T. cruzi involves mammalian hosts and the reduviid bug. Transmission to mammals, including humans, occurs with an insect bite to the skin, mucosal surfaces, or conjunctiva. The parasite subsequently replicates within host cells and ultimately ruptures these cells, liberating new parasites that invade local tissue. A cycle is established that involves an intermittent parasitemia with nonmultiplying infective forms that circulate in the blood. T. cruzi can be transmitted through blood transfusion.61 Vertical transmission of the parasite from mother to fetus has been reported. Congenital Chagas’ disease occurs in up to 2-5% of neonates born to infected women, and is associated with severe impairment in surviving infants and a high mortality rate.62
Clinical Spectrum. As mentioned above, Chagas’ disease may cause two syndromes, acute or chronic. With the exception of children, most patients with acute Chagas’ disease experience a mild clinical course. Acute Chagas’ disease involves the formation of inflammatory lesions at the site of entry, termed chagomas. The Romana sign is pathonogmonic of Chagas’ disease and is characterized by painless palpebral and periocular edema. Patients may complain of edema of the face and extremities, fever, malaise, and anorexia.58 Not uncommonly, invasion of the central nervous system occurs. In rare cases, T. cruzi may cause meningoencephalitis, a complication with a grim prognosis. Finally, a small number of patients develop acute myocarditis with death occurring secondary to congestive heart failure. Non-specific electrocardiographic changes may occur; however, the life-threatening arrhythmias characteristic of chronic Chagas’ disease are rare.32 Untreated, the acute illness spontaneously resolves in 4-6 weeks.
Following resolution of the initial illness, most patients enter the indeterminate phase of Chagas’ disease, characterized by lifelong, low-grade parasitemia and asymptomatic infection. Fewer than one-third of individuals subsequently will develop symptomatic chronic disease, which appears years following inoculation with the parasite. The heart is the most frequently afflicted organ in chronic Chagas’ disease; specimens of cardiac tissue reveal diffuse fibrosis and atrophy of myocardial cells. Myocardial damage eventually results in thinning of ventricular walls, apical aneurysms, biventricular enlargement, and conduction abnormalities. Common electrocardiographic abnormalities include right bundle-branch block, left anterior fascicular block, and complete atrioventricular block.63,64 Over time, patients develop symptoms reflecting dysrhythmias, cardiomyopathy, and thromboembolism. The cardiomyopathy most frequently affects the right ventricle, and many patients have signs of right-sided heart failure. Death typically occurs following fatal arrhythmias or congestive heart failure.
Some patients develop GI involvement, with dilatation of the esophagus and/or the colon. Symptoms include dysphagia, odynophagia, regurgitation, aspiration, abdominal pain, and chronic constipation.65 Patients with chagasic megacolon may develop acute obstruction and volvulus, leading to perforation, septicemia, and death.63,66 Although the pathogenesis of the cardiac and GI lesions of chronic Chagas’ disease is not well defined, it appears that local denervation may be involved.62
Diagnosis. The clinical findings of Chagas’ disease are nonspecific; consequently, serological studies may be required to establish the diagnosis. During acute infection, circulating forms of T. cruzi may be demonstrated by microscopic analysis of blood or on specialized culture medium. In contrast, diagnosis of chronic Chagas’ disease rests on detection of antibodies to parasite antigens. At this time, only one serological test for T. cruzi is available in the United States (Chagas’ IgG ELISA); the sensitivity and specificity of the assay have not yet been defined.62
Treatment. Current drug therapy for T. cruzi is unsatisfactory. Unlike the African Trypanosomal species, T. cruzi is not susceptible to most antiprotozoan medications. Benznidazole and nifurtimox have been shown to shorten the acute phase of the T. cruzi infection and reduce early mortality rates. The recommended dosages are: benznidazole at 5 mg/kg/day for 60 days and nifurtimox at 8-10 mg/kg/day for 90-120 days. Unfortunately, only 50% of treated patients achieve parasitologic cure.62 The efficacy of medical therapy in patients with chronic Chagas’ disease is less clear, but current recommendations do support the use of either nifurtimox or benznidazole regardless of clinical status or duration of disease.58 Cardiac transplantation has been attempted in patients with end-stage chagasic cardiomyopathy. Cure rates are dismal secondary to reinfection of the graft tissue.58
Dengue Fever. Epidemiology. Dengue is the most important arthropod-borne viral illness of public health concern in the world. Other arthropod-borne viral illnesses, including yellow fever, may cause higher morbidity and mortality rates; however, due to the rapidity with which their victims succumb from the disease, they seldom are seen outside their native region. Dengue is found in more than 100 countries, placing more than 2.5 billion people at risk. The disease causes a wide spectrum of clinical presentations, ranging from mild effects to severe hemorrhagic manifestations and shock, known as dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS).67 An estimated 50 million dengue infections occur worldwide each year, including 400,000 cases of DHF/DSS.68
Until 1981, only sporadic cases of DHF/DSS were reported in the Americas. Since then, the number of reported cases in Latin America and the Caribbean has increased steadily. In 2001, the WHO reported more than 250,000 cases of dengue and 3788 cases of DHF/DSS.69 In the United States, dengue outbreaks have been reported in communities along the Mexico-United States border. Movement of infected persons can bring the virus into dengue-free areas.70 Dengue also has been reported in non-border states. Between 1997 and 1998, the CDC reported 143 laboratory-confirmed cases of dengue, with the highest number of cases occurring in Florida and New York. Most of these cases originated in the Caribbean islands and Central America. It has been suggested that the true incidence of dengue in the United States actually is significantly higher than previously thought, as it is not a nationally reportable disease.71
Pathology. Dengue is caused by single-stranded RNA viruses belonging to the family Flaviviridae. Other flaviviruses include yellow fever, Japanese encephalitis, St. Louis encephalitis, and tick-borne viral encephalitides. Four species, or serotypes, have been recognized.67,72 Infection with one serotype confers type-specific life-long immunity but leaves the individual susceptible to other serotypes. Dengue is transmitted via the bite of an infected female Aedes aegypti mosquito. These mosquitoes can be found living between 30° north and 30° south of the equator, at elevations up to 2200 meters.73 The female mosquito tends to lay eggs in man-made containers, such as old automobile tires, flower vases, water-storage containers, and buckets. Infection commences with viral introduction via the bite of an infected mosquito. The virus replicates within cells of mononuclear phagocyte lineage and, after a 214-day incubation period, induces symptoms.
For unclear reasons, the degree of viremia can vary widely between individuals, causing significant ambiguity of clinical presentation.73 DHF/DSS occurs when an individual who was infected by one serotype encounters a second serotype. It is believed that pre-existent dengue antibodies enhance the infection and replication of a second virus in mononuclear cells. Consequently, the increased number of infected cells causes production of higher amounts of cytokines and chemical mediators, and a resultant clinical syndrome of vascular permeability, hypo-volemia, shock, and hemodynamic instability.74,75
Clinical Spectrum. Epidemiologically, dengue virus causes four diseases: nonspecific febrile illness, classic dengue fever, DHF, and DSS. Classic dengue is seen primarily in older children and adults and is characterized by an abrupt onset of symptoms. Younger children usually have a more benign course with minimal symptoms.73 High fevers, severe frontal and retro-orbital headache, myalgias, arthalgias, prostration, and anorexia are common complaints. On the third day of illness, a maculopapular rash appears on the chest, face, and flexor surfaces and lasts 48-72 hours. The febrile period of dengue usually lasts 5-7 days. Upon defervescence, the patient may experience an intense burning or itching sensation of the palmar and plantar surfaces. At this time, hemorrhagic manifestations also may appear, ranging from mild to life-threatening. Petechiae, purpura, epistaxis, metorrhagia, and gastrointestinal bleeding have been reported.73 A prolonged recovery period ensures, lasting weeks. It may be associated with weakness, fatigue, and depression.
Like classic dengue, DHF begins suddenly with high fever; however, DHF is distinguished by thrombocytopenia (< 105/mµL), capillary leakage induced hemoconcentration (20% increase over base hematocrit), hypoproteinemia, or polyserositis.73 Ultrasonography may reveal ascities and pleural or pericardial effusion. Hemorrhagic manifestations include petechiae, purpura, epistaxis, GI bleeding, and hematuria. The tourniquet test, which involves inflation of a blood pressure cuff to the patient’s median blood pressure for five minutes, is positive in more than 50% percent of patients.73 A positive test is indicated when three or more petechiae per square centimeter appear, although shock may underestimate the results. Aminotransferases may be elevated, and leukopenia and lymphocytosis are not uncommon.73 Disturbances in the coagulation cascade may cause prolongation of prothrombin (PT) and partial thromboplastin times (PTT), decreased fibrinogen levels and increased fibrin degradation products.
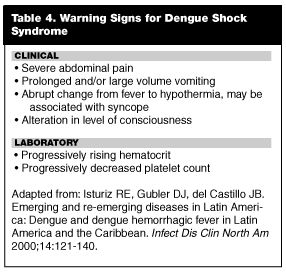
The criteria for DSS include signs of hemodynamic instability, including hypotension and narrowing of pulse pressure (< 20 mmHg). Warning signs for DSS have been described and are listed in Table 4. Circulatory failure may develop quickly and progress to overt shock and death within 12-24 hours. Death tends to occur on the fourth or fifth days of illness. Brief episodes of shock with DSS can be well tolerated, but prolonged shock carries a poor prognosis. The laboratory abnormalities for DSS are similar to that of DHF but are more severe. Unlike classic dengue, the recovery period following the acute illness is rapid and uneventful.
Diagnosis. Laboratory diagnosis of dengue can be accomplished via demonstration of either the virus or antiviral antibodies in serum. Both polymerase chain reaction (PCR) and immunofluorescence assays are available and will document either the presence of IgM antibodies or a rise in IgG antibody titers in paired samples collected during the acute and convalescent phase of the illness. The timing of appearance of IgM antibodies varies considerably among patients, but 90% will have detectable antibodies by the fifth day after onset of symptoms.69 Measurement of IgG antibodies is less clinically useful than IgM antibodies and is usually of epidemiologic interest only.
Treatment. Management of dengue fever, DHF, and DSS is primarily supportive. Patients with dengue require adequate fluid resuscitation and maintenance, analgesia, and antipyretics. Aspirin and nonsteroidal anti-inflammatory drugs should be avoided, as they may exacerbate platelet dysfunction and GI mucosal toxicity.69 Patients with dengue do not universally require hospitalization, but all should be followed closely. Patients with evidence of DHF/DSS or those with co-morbidities should be admitted, preferably to an intensive care unit. The WHO has created guidelines for the treatment of DHF/DSS, tailored primarily for smaller hospitals.76 The most important elements in managing a patient with DHF/DSS are judicious monitoring and supportive treatment of blood pressure, hematocrit, platelet count, urinary output, hemorrhagic manifestations, and level of consciousness.69 Fluid replacement should be aggressive enough to maintain effective circulation without aggravating plasma leakage into serous and interstitial spaces.
Asia and South Asia
Hemoglobin Disorders. Epidemiology. Anemia is common in immigrants from Asia, Southeast Asia (Vietnam, Laos, Cambodia), and South Asia (India, Pakistan). During the past 30 years, several million Asian refugees have entered the United States, largely due to war in Vietnam and ongoing political chaos in Southeast Asia.77 As a result, the various hematological abnormalities common to Asian immigrants are becoming more common in the ED. The majority of the red blood cell (RBC) disorders are not associated with clinical significant disease. However, some may cause intrauterine fetal demise or chronic hemolytic anemia requiring lifelong RBC transfusions.77
By far, the most common cause of anemia in Asian immigrants is iron deficiency. One large study of Southeast Asian children who had recently emigrated to the United States found that 48% of Cambodian children, 19% of Laotian children, and 24% of Vietnamese children demonstrated anemia.78 Dietary habits and parasites were the most common culprits. In 37% of children, stool analysis identified hookworm.78 For a more detailed discussion on intestinal parasites, see Part I of this series. (Emerg Med Rep 24;4:31-50.)
It is becoming increasingly apparent that a significant proportion of Asian immigrants may have a hemoglobinopathy. Notwithstanding, iron-deficiency anemia should be diagnosed and treated prior to performing hemoglobin electrophoresis. One study of Southeast Asian refugees found that 40% of all immigrants had evidence of at least one major red blood cell abnormality; 22% had glucose-6-phosphate dehydrogenase deficiency, 16% had hemoglobin E disease, 6% had alpha-thalassemia, and 4% had beta-thalassemia.77 Another study of recent immigrants from Southeast Asia noted that 241 of 778 had microcytosis; of these, only 31% were iron-deficient and the rest were shown to have a hemoglobinopathy.79 Hence, microcytosis and anemia in individuals from Asia can stem from a multitude of causes, including malnutrition, parasite infection, or hemoglobinopathy. Treatment varies according to the etiology.
Glucose-6-Phosphate Dehydrogenase Deficiency. The EP occasionally will encounter an immigrant who appears to have a new hemolytic anemia. Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is an X-linked recessive disorder that affects millions of people worldwide, particularly in Mediterranean countries, Africa, and Asia.77 G6PD deficiency is the most common RBC enzyme disorder to induce hemolysis. It occurs with surprising frequency in Laotian and Cambodian immigrants; various studies have demonstrated a prevalence of 10-30%.77 Of note, Asian individuals with G6PD deficiency usually have a milder form of the disease, and hemolysis in this cohort only occurs with infection or exposure to certain oxidant drugs.77 Most of the antimalarial drugs will precipitate hemolysis in individuals with the disease.77
Thalassemia. Thalassemia is a disease of RBCs, in which a variety of gene mutations results in defective synthesis of either the alpha- or beta-globin chains of hemoglobin.80 Historically and evolutionarily, the disease occurred selectively in areas where malaria was endemic, namely the Mediterranean, the Middle East, Southeast Asia, and the Indian subcontinent.81 The prevalence of thalassemia in these regions has been attributed to a protective effect of thalassemia trait against malaria infection.81 Until the 1970s, most cases of thalassemia in the United States occurred in individuals of Italian or Greek ethnicity.82 However, immigration of the past two decades has resulted in a changing profile of the disease; the number of new reported cases has declined in the Italian and Greek populations, while there has been a steady increase in the number of non-Mediterranean patients diagnosed with the disease.82 A recent survey of 45 pediatric hematology centers in the United States noted that 23% of beta-thalassemia cases now occur in Asian, Southeast Asian, or South Asian patients.82
The thalassemias are characterized by decreased or absent production of one of the globin chains that comprise a normal hemoglobin molecule.77 Each syndrome is named according to the globin chain whose synthesis is decreased. alpha-thalassemia results from decreased production of alpha-chain globins; beta-thalassemia results from decreased production of beta-chain globins. Symptoms of the disease result from reduced production of functional hemoglobin, and, more importantly, from accumulation of unpaired normal globin chains.80,81
beta-Thalassemia. Homozygous thalassemia (thalassemia major or Cooley’s disease) occurs when both genes involved in the synthesis of beta-chains are impaired. It is a severe disease marked by a massive hemolytic anemia that is fatal in childhood if untreated.81,82 The child usually becomes symptomatic at 6 months of age as levels of fetal hemoglobin decrease.83 Common presenting symptoms include pallor, irritability, growth retardation, abdominal swelling secondary to hepatosplenomegaly, and jaundice.83 The EP can expect these children to present in extremis due to the effects of severe anemia, with congestive heart failure or prostration. Laboratory studies reveal hemoglobin levels in the range of 3-7 g/dL with a decreased reticulocyte count and significant microcytosis (MCV= 50-60).83 Without chronic transfusions, 80% of children with thalassemia major will die before reaching 5 years of age.81
The current mainstay of thalassemia major treatment consists of frequent blood transfusions designed to keep hemoglobin levels in the range of 9-10 g/dL.84,85 Unfortunately, chronic transfusion therapy results in hemosiderosis and symptoms of iron overload. Without chelation therapy, all children with thalassemia major develop myocardial and hepatic hemosiderosis by the second decade of life, with death usually occurring at the end of the second decade.80,85 Affected individuals will demonstrate portal hypertension, or frank cirrhosis, accompanied by the protean manifestations of liver disease, including coagulopathy, bleeding varices, and ascites. Concurrent viral hepatitis is a common problem. Cardiac hemosiderosis results in the gradual development of refractory congestive heart failure. Death due to cardiac disease usually occurs by the third decade of life and typically is secondary to arrhythmia or pump failure.83,85 While the introduction of chelation therapy with deferoxamine mesylate has revolutionized the treatment of thalassemia, this intervention has only become standard in the past 15 years and remains largely unavailable to children of the developing world.80
Occasionally, a patient may present requesting a blood transfusion; individuals typically require 1-3 units of packed RBCs every 3-5 weeks.84,85 Deferoxamine therapy now is given as a 12-hour subcutaneous infusion that the patient self-administers overnight.85,86,93 It may be associated with local infections or hematomas and more chronic effects, including impaired sight and hearing.84 Consultation with a hematologist is helpful in these situations. Patients who require yearly transfusions exceeding 200 mL RBCs per kilogram of body weight may benefit from splenectomy. Removal of the spleen has been shown to reduce transfusion requirements and concomitant iron accumulation, but the procedure places the patient at particularly high risk for post-splenectomy sepsis.85 Pneumococccus sepsis is most common and presents with a fulminant course that carries a 10-30% mortality rate. Splenectomized patients may be maintained on prophylactic penicillin and should be offered vaccination against Pneumococcus (Pneumovax).
alpha-Thalassemia. alpha-thalassemia is a global disease, occurring in Africa, the Mediterranean basin, and throughout Asia. In fact, alpha-thalassemia is presumed to be the most common genetic disorder in the world.87 Individuals of Southeast Asian descent most commonly are affected, with a prevalence of 12-14% in Cambodians and Laotians and 8% in Vietnamese individuals.78 alpha-globin polypeptide production is controlled by a total of four genes.
Patients who have three alpha-globin deletions suffer from hemoglobin H disease. An estimated 13,000-16,000 infants are born each year in Southeast Asia with hemoglobin H disease.88 During the neonatal period, affected individuals have marked hypochromia and microcytosis; beyond infancy, the inequality in globin chain synthesis is coupled with beta-globin excess and the occurrence of hemoglobin H. Such patients have a mild to moderate hemolytic anemia with hemoglobin levels that range from 6-12 g/dL.77 Fatigue and splenomegaly are common, but patients rarely require blood transfusions.77 Life expectancy is normal. Splenectomy rarely is necessary but may be essential in patients with severe anemia and hypersplenism.77 Hemoglobin H patients should be instructed to avoid oxidant drugs that can accelerate hemolysis. Alternately, alpha-thalassemia major (hydrops fetalis or Bart’s disease) occurs when all four genes necessary for alpha-globin production are mutated and there is a total lack of alpha-globin production.77 This condition is incompatible with life; most infants are stillborn or die shortly after birth.77,87 The neonate will demonstrate a severe anemia with marked anasarc.87 A negative Coombs’ test excludes blood group incompatibility.77,87
Liver Flukes. Epidemiology. The liver flukes are trematode worms endemic in many areas of the Far East, specifically China, Japan, Korea, Hong Kong, Taiwan, Vietnam, Thailand, Laos, and Cambodia.89 The two main species of liver fluke that infect humans are Clonorchis sinesis (Chinese liver fluke) and Opisthorchis viverini.90 Both species produce disease by invading the biliary tree and, after years of infection, produce symptoms though chronic hepatic fibrotic changes.90 In certain regions of Hong Kong, the prevalence of liver fluke infection in the general population reaches 80%.91 In the United States, the disease is purely an imported infection.90 Liver fluke disease should be considered in the differential of Asian immigrants who complain of right upper quadrant pain or cholangitis as untreated disease causes chronic complications that may be fatal.
Life Cycle. Humans acquire liver fluke infection by consuming raw, smoked, pickled, or undercooked fish.90 Parasite eggs pass from the feces of infected humans into fresh water, where they hatch into larvae that invade snails; after a maturation phase, the larvae leave the snail and proceed to infect certain strains of freshwater fish.89,90 The liver fluke larvae migrate to muscles and other organs of the fish, where they encyst and may remain viable for years.89,90 When a person consumes infected fish, the larvae decyst in the intestine.89,90 They then migrate through the Ampulla of Vater into the bile duct, where they remain for the rest of their lives.89 After three weeks, the mature worms begin to lay eggs that are passed with bile into the stool. Mature worms live, on average, 8-10 years but may survive as many as 30 years in the biliary tract.89,90
Clinical Spectrum. More than two-thirds of individuals infected with liver fluke remain asymptomatic.89,90 Patients with heavier infections, however, develop inflammation and fibrosis of the biliary tract, due to both mechanical irritation and an immunogenic reaction to egg antigens.91 The result is a syndrome of intermittent biliary obstruction that resembles repetitive cholecystitis or cholangitis.91 The patient presents with right upper quadrant pain, jaundice, tender hepatomegaly, anorexia, and weight loss and frequently will relate a history of multiple prior similar episodes. Lab tests may demonstrate a mildly elevated bilirubin and alkaline phosphatase.91 In the setting of acute obstruction, transaminases also may be increased. Eosinophilia, however, is rare.91 Gallstone formation is common and contributes to obstructive symptoms. Some patients develop recurrent pyogenic cholangitis associated with fever, right upper quadrant pain, sepsis, and even liver failure.89,92,93 This complication is believed to be secondary to bacterial superinfection in the setting of fibrosis of the biliary tree. Adult worms may migrate into the pancreatic duct and cause acute pancreatitis.93 Chronic liver fluke infection has been associated with an increased risk of cholangiocarcinoma.89,93
Diagnosis. An ultrasound of the right upper quadrant frequently will be the first diagnostic study that hints at the diagnosis of liver fluke disease. Patients with chronic liver fluke infection will demonstrate a dilated common bile duct and often have cholelithiasis.91,93 CT of the abdomen will show similar findings. Definitive diagnosis is accomplished by finding parasite ova in the patient’s stool.89 Ova also can be identified in bile samples obtained during surgery.89 Serological assays for liver fluke infection are available but not commonly used as stool diagnosis is highly sensitive.9
Treatment. Conservative management of liver fluke infection usually is effective. High cure rates are obtained via praziquantel 25 mg/kg three times a day for 1-2 days.91,95 Albendazole 10 mg/kg twice a day for 7 days91,95 is an acceptable, albeit more prolonged, alternative. Patients with acute obstruction will require decompression and drainage in addition to anti-parasitic therapy: options include endoscopic retrograde cholangiopancreatography (ERCP), cholecystectomy, or cholecystostomy.89,92 Patients with evidence of systemic infection should receive parenteral antibiotics with broad gram-negative bacterial coverage. Unfortunately, prognosis is dismal for individuals with heavy infections. Death occurs from gram-negative sepsis or cholangiocarcinoma.91,95
Africa
Schistosomiasis. Epidemiology. Schistosomiasis, also referred to as "Bilharzia" in Africa, is an infection caused by trematode blood flukes of the family Schistosoma.95,96 Schistosomiasis is second only to malaria as the most common parasitic disease of humans. The disease is estimated to affect 200 million people worldwide, predominantly in Africa, Asia, and South America, and causes 200,000 deaths annually.95-98 In the United States, approximately 400,000 individuals have chronic Schistosomiasis, due to travel and immigration.98
Schistosomal disease in humans is caused by one of five species: S. haematobium, S. mansoni, S. japonicum, S. mekongi, and S. intercalatum.97 Most infections with S. haematobium, S. mansoni, and S. intercalatum species occur in sub-Saharan Africa.74 S. japonicum is found primarily in China, Indonesia, and the Philippines; S. mekongi is seen in Cambodia and Laos.96 All countries in which schistosomal disease is endemic are located within 36° north or south of the equator, where local climate conditions are ideal for the snail that serves as an intermediate host to the parasite.98 Although most patients with schistosomiasis in the United States are immigrants from Africa, the EP should be aware that the disease also is prevalent in specific regions of Asia.
Life Cycle. All species of Schistosoma share a common life cycle. Parasite eggs are excreted by a human carrier into fresh water where they hatch and invade host snails. Within the snails, the parasites mature into a larval form.97 The larvae exit the snail and swim in search of a human host; if no host is found within 48-72 hours, the larvae die.98 Upon encountering a human, the larvae penetrate any exposed skin and migrate through the venous system toward the lungs.98 Eventually, the schistosoma larvae make their way to the left side of the heart and are carried by arterial blood to finally arrive in the portal hepatic circulation.97 Upon reaching the liver, they travel against portal blood flow into various mesenteric venous plexuses. The exact anatomic site of migration is species-specific: S. haemotobium travels to the venous plexus of the bladder, whereas S. japonicum and S. mekongi migrate to the superior mesenteric vein.98 S. mansoni inhabits the inferior mesenteric vein.98 After a period of 4-6 weeks, the mature schistosome begins to produce eggs that penetrate the wall of the intestine or bladder and are passed into stool or urine. Alternately, some eggs travel from their venous plexuses to the portal circulation of the liver and lodge in small tributaries of the portal vein.
Most of the signs and symptoms of schistosomiasis occur as a result of the human host’s response to egg deposition.96,98 Adult worms are not recognized by the immune system, provoke minimal immunological response, and can live unharmed in host tissue for their average life cycle of five years.95,98 It appears that the adult worms shed surface antigens and acquire the host’s blood group glycolipids and major histocompatibility complex (MHC) antigens. Expression of these host antigens on their surface permits the parasites to simulate host tissue and avoid attack by the immune system.98 By contrast, schistosomal eggs provoke a strong immunological reaction. Acutely, the eggs trigger a hypersensitivity response; long-term, egg deposition results in a granulomatous reaction and eventual fibrosis of the human tissue surrounding the eggs.96,98
Clinical Spectrum. Acute schistosomal disease, or Katayama fever, occurs 4-6 weeks after infection and coincides with the onset of egg deposition.99 Symptoms are believed to be secondary to a hypersensitivity response to egg antigens.99 The patient develops fever, chills, arthralgias, myalgias, cough, wheezing, abdominal pain, diarrhea, and headache. Hepatomegaly, splenomegaly, and lymphadenopathy are the primary physical findings. A diagnostic clue is the presence of peripheral eosinophilia in an individual who recently has traveled to an endemic region.99 At this stage, stool and urine studies frequently are negative but the schistosomal serological assay is positive.97 Untreated, Katayama fever usually resolves spontaneously after several weeks with minimal morbidity.96,98,99 The treatment is the same as for chronic schistosomiasis infection.96,98,99
In the United States, patients infected with Schistosomiasis more commonly will present to the ED with the manifestations of chronic disease. In this setting, pathology occurs secondary to fibrosis and scarring of the urinary tract, GI tract, or liver. In genitourinary schistosomiasis, hematuria is an early finding.96 Ureteral obstruction with concomitant hydronephrosis and renal colic occur later.96 Bacterial superinfection is common. Schistosomal infection also appears to play a role in the development of squamous cell bladder cancer, although its exact mechanism remains unclear.96 In Egypt, squamous cell bladder cancer represents 18-28% of all documented cancers, occurring at a rate of 11 per 100,000 people.96 Women with genitourinary schistosomiasis may develop ulcers or wart-like lesions of the vulva that resemble papillomatous disease.96
Intestinal schistosomiasis tends to be one of the milder forms of disease and primarily affects the large bowel.98 The individual develops mucoid bloody diarrhea, tenesmus, and abdominal cramps.96,98 Eventually, colonic strictures may cause large bowel obstruction or rectal prolapse; fistulas and intussusception also may occur.96 Contractile dysfunction results in obstipation. On exam, the patient has guaiac positive stools with an iron-deficiency anemia and a peripheral eosinophilia.96,98 These patients do not appear to be at increased risk of colorectal carcinoma. Hepatosplenic schistosomiasis occurs in 4-8% of individuals with chronic infection and accounts for the majority of morbidity from the disease.96 After many years of infection, peri-portal fibrosis results in portal hypertension. The patient develops hepatomegaly, splenomegaly, and esophageal varices.96 Of note, unless there is concurrent infection with viral hepatitis, hepatocellular function remains preserved.98 Transaminases usually are normal, and alkaline phosphatase may be mildly elevated.96 While striking hepatomegaly and ascites can occur in chronic schistosomiasis, other stigmata of cirrhosis, such as testicular atrophy, jaundice, and spider nevi, are uncommon. Death results from exsanguination from bleeding varices.95 The exact relation between schistosomiasis and hepatocellular carcinoma is uncertain, although there is some evidence that S. japonicum represents a risk factor for this cancer.96,98
Diagnosis. The gold standard for diagnosis of schistosomiasis is detection of eggs in stool or urine.95 Multiple samples may be required; a single stool specimen has a sensitivity of 50-70% that approaches 90% when three samples are examined.98,100 The species of Schistosoma easily are distinguished based on the morphology of their eggs.95 Abdominal ultrasound is useful for measuring spleen and liver size, assessing the degree of fibrosis, and detecting the complications of portal hypertension. More recently, ultrasound has been employed for monitoring the response to therapy.98
Serological assays for detection of antibodies to Schistosoma are available, but are of limited value in immigrants because they remain positive for life,95 and individuals from endemic areas can be expected to have high levels of seropositivity due to exposure at an early age.98 This contrasts with United States natives, who usually have negative serology at baseline, and in whom a positive assay usually will reflect recent disease. More recently, an enzyme-linked immunosorbent assay (ELISA) for detection of schistosome antigen has been developed; preliminary studies suggest a sensitivity of 95% and a specificity of 100%.98,101,102 The clinical utility of the test still is to be determined.
Treatment. The drug of choice for treatment of schistosomiasis is praziquantel administered as 40 mg/kg in 2-3 divided doses during one day.96 A slightly higher dose of 60 mg/kg is required for S. japonicum infection.96 Typically, 60-90% of patients will be cured with a single treatment.96 Re-examination of stool or urine is recommended at three months and six months post-treatment; if any eggs are found, the patient should be re-treated with praziquantel at the same dose.96 Although advanced organ fibrosis usually is unchanged, moderate disease of the liver and urinary tract may improve with anti-parasitic therapy. There have been several reports of drug resistance in Egypt and Africa, but it appears that most patients can receive multiple courses of praziquantel for recurrent infections with good cure rates.98
Patients with Katayama fever should receive standard doses of praziquantel that may be supplemented with low dose steroids if symptoms are severe.95,99 Variceal complications may require any or all of the following: endoscopic sclerotherapy, transjugular intrahepatic portosystemic shunts (TIPS), and propranolol prophylaxis.
Female Circumcision. Epidemiology. Female circumcision, also referred to as "female genital mutilation" or "female genital cutting," is a surgical modification of the external female genitalia.103 The procedure is practiced primarily in 28 countries in Africa, with sporadic occurrences in the Middle East and Asia.103 According to the WHO, female circumcision typically is "associated with poverty, illiteracy, and low status of women" and occurs in some of the poorest countries in the world.104 While the procedure traditionally has been associated with the Muslim faith, it also is practiced in Africa by Christian and Jewish societies.105 The prevalence of female circumcision in Africa ranges widely with a few countries containing the vast majority of cases; various surveys have indicated that 98% of Somali women and 70-97% of Egyptian women undergo some form of surgical genital alteration.106-108
In the United States, the EP can expect to encounter female circumcision primarily in women who have emigrated from Somalia, Ethiopia, Eritrea, or the Sudan.103,105 These countries are, coincidentally, the regions where the most severe forms of female genital alteration are practiced.106,109 Although there has been no formal census in the United States, preliminary studies suggest that approximately 27,000 immigrant women in Sweden and 30,000 immigrant women in Italy have undergone female circumcision.103 As the number of African immigrants to the United States continues to increase, it is important to understand the myriad medical and psychological side effects that accompany female circumcision.
Classification. In Africa, female circumcision usually is done between 4 and 10 years of age.109,110 There is some evidence that, when performed in the United States, the procedure occurs at an earlier age, probably due in part to parents’ fear of reprisal. As practiced in an immigrant’s home country, female circumcision usually is performed by a local midwife or layperson without anesthesia and involves the use of a variety of instruments, including knives, razor blades, broken glass, and scissors.104,109 Physicians perform approximately 12% of surgeries, usually for upper-class families living in larger cities. There is scant data on how the procedure occurs in developed nations, where it is almost universally illegal.
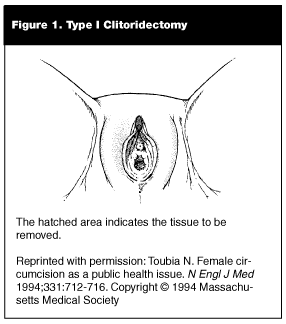
Female circumcision generally can be categorized into three classes based on the severity of surgical modification. Type I circumcision also is referred to as clitoridectomy; in this procedure, part or all, of the clitoris is removed, and the remaining external genitalia is left intact.104,105 (See Figure 1.) Type I circumcision commonly is referred to as "sunna" circumcision, meaning "following the Prophet’s tradition."104,105
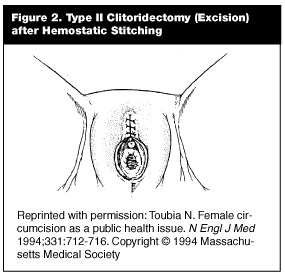
Type II circumcision, or excision, involves removal of the entire clitoris and some or all of the labia minora.103,104,110(See Figure 2.) After surgical removal of the labia, bleeding from the clitoral artery is tamponaded by a variety of methods, including sutures of catgut, thorns, or mud poultices.110
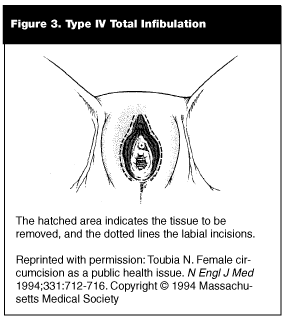
Types III and Type IV circumcision (or pharaonic circumcision) usually are referred to as infibulation and represent the most severe forms of the procedure.103,105,109,110 (See Figure 3.) In this practice, all of the clitoris and labia minora are excised. Incisions then are made in the labia major to create raw surfaces, which are stitched together to cover the urethra and vaginal introitus, leaving only a small posterior opening for urinary and menstrual blood flow.105,109,110 After the surgery, the girl’s legs may be bound together for up to 40 days to ensure formation of adequate scar tissue.104
Female circumcision is best understood as a phenomenon of poorer countries in which the main source of social and economic security for a woman is marriage.103,110 Anthropologists have described it as a physical marking of a woman’s appropriateness for marriage and a way of ensuring her chasteness and virginity.104,105,110 The procedure also is thought by some Muslims to be a requirement of their faith, although multiple Islamic scholars and theologians have disputed this theory, and there are many major Muslim countries that do not practice female circumcision.105,109 Upon arrival in a new country such as the United States, preservation of cultural identity becomes even more important to the immigrant, who is isolated in a different and often condemning society.110 Some families may fear that no men will marry their daughters if the procedure is not performed.104,111 For these reasons, female circumcision continues to occur in developed countries among the children of immigrants.110 There are, unfortunately, case reports of female circumcision that have been performed throughout Europe that have resulted in deaths of the children.104,113
Health Consequences. The immediate medical consequences of female circumcision include local infection, septicemia, tetanus, and gangrene.105,106 Exsanguination probably is the most significant immediate danger.111 Complications are more common in the Third World secondary to lack of sterility, but female circumcision performed in developed countries is illegal and may be subject to some of the same poor conditions. There is real concern for the potential transmission of blood-born pathogens such as hepatitis and HIV.103
The more chronic side effects of female circumcision usually occur with infibulation (Types III and IV) and result from interference with the drainage of urine and menstrual blood.110 A tightly infibulated woman requires 10-15 minutes to urinate (drop by drop) and usually has multiple, chronic urinary tract infections, nephrolithiasis, and renal damage.104,110 Chronic pelvic infection is common, resulting in pelvic and back pain, dysmenorrhea, and sometimes infertility.104-110 Menstruation may last for more than 10 days and often is malodorous, causing some women significant anxiety.104
One of the most common complications of female circumcision is the formation of dermoid cysts in the line of the scar.105,110 These cysts results from epithelial cells and sebaceous glands that are embedded within the scar; they may be as small as a pea or as large as a grapefruit.110 Keloid formation also may occur, and stitch neuromas may form from entrapment of a nerve ending within the scar.105-110 Sexual intercourse may be difficult or even impossible in tightly infibulated women. Penetration requires either a midwife to cut the scar on a woman’s wedding night (deinfibulation)103,108 or the husband gradually may enlarge the opening over the course of several weeks using his fingers, a razor, or a knife.104 Such an event can result in severe dyspareunia.105
Perhaps the most dangerous complications occur with childbirth. For the fetal head to exit, the infibulation scar must be enlarged by anterior episiotomy.104,110 American physicians unfamiliar with the effects of female circumcision may not understand how crucially important this is, as noted by one family medicine doctor who assisted a Somalian woman through her labor in the United States.108 If the infibulation scar is not enlarged prophylactically, the result is either severe perineal tears or fetal demise.110 Furthermore, if the fetal head exerts pressure for a prolonged period of time on the septum between the vagina and bladder, a vesicovaginal fistula may form, with resultant urinary incontinence.104,110
Management. EPs should expect that the speculum exam will be difficult in patients who have undergone Types II-IV circumcision. Using a pediatric speculum may aid in the exam, and the bimanual portion may be performed using a single finger and a rectal exam.104,109 These women are at increased risk for pelvic infection. Therefore, if a reliable physical examination cannot be performed, they should be treated presumptively in the face of appropriate symptoms. The same is true for patients with urinary complaints.
During childbirth, it has been recommended that women with Types III and IV infibulation receive an anterior episiotomy during the second stage of labor.104,109 Good preparation may avoid the need for a large posterior episiotomy.104
Legal Issues. In 1982, the WHO stated that it, " has consistently and unequivocally advised that female circumcision should not be practiced by any health professionals in any setting."104 Since 1985, the procedure has been illegal in Sweden and the United Kingdom.105,109,110 No formal law exists in France, but several cases of female circumcision have been prosecuted successfully as child abuse in the courts.109,110 The international medical community, including the World Medical Association, the American Academy of Pediatrics, and the International Federation of Gynecology and Obstetrics, has issued statements condemning the procedure as medically unnecessary and potentially life-threatening.109
Childhood female circumcision became illegal in the United States in 1995 with the Federal Prohibition of Female Genital Mutilation Act.108,109 This law prohibits, as child abuse, the procedure on girls younger than 18 years of age.108 Occasionally, adult women who have undergone deinfibulation for childbirth may request reinfibulation following successful delivery.104,108 There is no current United States law regarding the infibulation of consenting adult women, but there is a strong possibility that reinfibulation would be considered illegal in a United States court of law.108,110
Summary
The slogan of the environmental movement, "Think globally, act locally," is an appropriate mantra for physicians caring for today’s immigrants and refugees. Due to immigration and travel, once localized microbes now are able to reach the four corners of the globe at jet speed and, consequently, clinicians are realizing they may be facing exotic possibilities when evaluating their patients’ illnesses. Growing numbers of immigrants are arriving from tropical and underdeveloped countries, bringing with them increasingly diverse diseases. Many of these illnesses have nonspecific presentations and may mimic common temperate maladies. Within the United States, once nearly eradicated health problems are re-emerging among immigrants at a disquieting rate. These include tuberculosis, chronic hepatitis B, intestinal parasites, measles, and nutritional deficiencies. In contrast to their predecessors of the early 20th century, the new immigrants of the 1980s and 1990s have had more difficulty creating de novo social support networks.2 Many immigrants originating from Africa, Eastern Europe, the Middle East, and other politically unstable areas have been victims of physical torture or abuse. Another rapidly expanding group in the United States is undocumented illegal aliens. They have few, if any, options for health care. The current climate of anti-immigrant sentiment and associated social and economic finger-pointing has generated an alarming potential for punitive laws directed towards émigrés, both legal and illegal. At risk is their access to health care. Thus, it is likely that immigrants increasingly will access the ED. The EP may provide comprehensive, compassionate care through careful history taking and physical examination with respect for the immigrants’ physical, cultural, and educational beliefs, and by appreciation of the special physical and mental health problems encountered in these patient populations.
References
1. Kennedy JF. A Nation of Immigrants. New York; Harper Perennial Library: 1986.
2. Markel H. Caring for the foreign born. The health of immigrant children in the United States, 1890-1925. Arch Pediatr Adolesc Med 1998;152:1020-1027.
3. Walker P, Jaranson J. Refugee and immigrant health care. Med Clin North Am 1999:4:1103-1120.
4. Lillie-Blanton M, Hudman J. Untangling the web: Race/ethnicity, immigration, and the nation’s health. Am J Pub Health 2001;91:1736-1738.
5. Singh G, Siahpush M. All-cause and cause-specific mortality of immigrants and native born in the United States. Am J Pub Health 2001;91:392-399.
6. Evans C. Immigrants and health care: Mounting problems. Ann Intern Med 1995;122:309-310.
7. Brillman J, Quenzer R. Infectious Disease in Emergency Medicine. Philadelphia; Lippincott-Raven Publishers; 1998:409-425.
8. Hoffman S. Tropical medicine and the acute abdomen. Emerg Med Clin North Am 1989;7:591-609.
9. Garcia H, Del Brutto O. Taenia Solium cysticercosis. Emerging and re-emerging diseases in Latin America. Infect Dis Clin North Am 2000;14:97-116.
10. Garg R. Neurocysticercosis. Postgrad Med J 1998; 4: 321-326.
11. Yamashita P, Kelsey J, Henderson S. Subcutaneous cysticercosis. J Emerg Med 1998;16:583-586.
12. Brown WJ, Voge M. Cysticercosis: An Update. Rev Infect Dis 1988;10:1075-1087.
13. Schontz PM, Santi E, Plancarte A, et al. Community-based epidemiologic investigation of cysticercosis due to Taenia Solium: Comparison of serological screening tests and clinical findings in two populations in Mexico. Clin Inf Dis 1994; 18:879-885.
14. Stephenson J. Health at home means watching the global village. JAMA 1995;273:1648-1649.
15. Schantz PM, Moore AC, Munoz JL, et al. Neurocysticercosis in an Orthodox Jewish Community in New York City. N Engl J Med 1992;327:692-695.
16. Coyle C, Wittner M, Tanowitz H. Cysticercosis. In: Guerrant RL, Walker DH, Weller PF, eds. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia; Churchill Livingstone;1999:993-999.
17. Jong E. Intestinal and extraintestinal tapeworms, including Echinococcus and Cysticercus. In: Jong E, ed. The Travel and Tropical Medicine Manual. Philadelphia; W.B. Saunders Co.;1987:453-459.
18. Bern C, Garcia HH, Evans C, et al. Magnitude of the disease burden from neurocysticercosis in a developing country. Clin Infect Dis 1999;29:1203-1209.
19. Nash T, Pretell J, Garcia H. Calcified cysticerci provoke perilesional edema and seizures. Clin Infect Dis 2001;33: 1649-1653.
20. Moran GJ, Kyriacou DN, Newdow MA, et al. Emergency department sentinel surveillance for emerging infectious diseases. Ann Emerg Med 1995;26:351-354.
21. Kitchen L. Case studies in international medicine. Am Fam Physician 1999;59:3040-3044.
22. Brutto OH, Rajshekhar V, White AC Jr., et al. Proposed diagnostic criteria for neurocysticercosis. Neurology 2001;57: 177-183.
23. Desai SB, Shah VC, Tavri DJ, et al. Neurocysticercosis: Comparison of CT scan and MRI. Study of 35 cases. Neuroradiology 1991;33 (suppl):578-579.
24. Martinez HR, Rangel-Guerra Ra, Elizondo G, et al. MR imaging in neurocysticercosis: A study of 56 cases. Am J Neuroradiol 1989;10:1011-1019.
25. Gilman RH, Del Brutto OH, Garcia HH, et al. Prevalence of taeniosis among patients with neurocysticercosis is related to severity of infection. Neurology 2000;55:1062.
26. Tsang VCW, Brand JA, Boyer AK. An enzyme linked immunoelectrotransfer blot assay and glycoprotein antigens for diagnosing human cysticercosis (Taenia solium). J Infect Dis 1989;159:50-59.
27. Wilson M, Bryan RT, Friend JA, et al. Clinical evaluation of the cysticercosis by enzyme linked immunotransfer blot in patients with neurocysticercosis. J Infect Dis 1991;164: 1007-1009.
28. Mitchell WG, Crawford TO. Intraparenchymal cerebral cysticercosis in children: Diagnosis and treatment. Pediatrics 1988;82:76-82.
29. Del Brutto OH, Santibanez R, Noboa CA, et al. Epilepsy due to neurocysticercosis: Analysis of 203 patients. Neurology 1992;42:389-392.
30. Vazquez V, Sotelo J. The course of seizures after treatment for cerebral cysticercosis. N Engl J Med 1992;327:696-701.
31. Carpio A, Santillan F, Leon P, et al. Is the course of neurocysticercosis modified by treatment with anthelmic agents? Arch Intern Med 1995;155:1982-1988.
32. Padma MV, Behari M, Misra NK, et al. Albendazole in neurocysticercosis. Natl Med J India 1995;8:255-258.
33. Kramer LD. Medical treatment of cysticercosis: Ineffective. Arch Neurol 1995;52:101-102.
34. Escobedo F, Penagos P, Rodriguez J, et al. Albendazole therapy for neurocysticercosis. Arch Intern Med 1987;147: 738-741.
35. Del Brutto OH, Sotelo J, Aguirre R, et al. Albendazole therapy for giant subarachnoid cysticerci. Arch Neurol 1992;49: 545-547.
36. Takayanagui OM, Jardim E. Therapy for neurocysticercosis: Comparison between praziquantel and albendazole. Arch Neurol 1992;49:535-537.
37. Cruz I, Cruz ME, Carrasco F, et al. Neurocysticercosis: Optimal dose treatment with albendazole. J Neurol Sci 1995;133: 152-154.
38. Wadia NH. Neurocysticercosis. Tropical Neurology. London: WB Saunders, 1995:247-273.
39. Wadia NH, Srinivas D, Bhatt M. Disseminated cysticercosis. New observations, including CT findings and experience with treatment by praziquantel. Brain 1988;111:597-614.
41. Proano JV, Madrazo I, Avelar F, et al. Medical therapy for neurocysticercosis characterized by giant subarachnoid cysts. New Engl J Med 2001;345:879-884.
41. Hutcheson K III, Kalafian M, Taylor I, et al. Neurocysticercosis. South Med J 2000;93:666-668.
42. Petri WA, Singh U. Diagnosis and management of amebiasis. Clin Inf Dis 1999;29:1117-1125.
43. Aucott JN, Ravdin JI. Amebiasis and "nonpathogenic" intestinal protozoa. Infect Dis Cin North Am 1993;7:467-485.
44. Hoffner RJ, Kilaghbian T, Esekogwu VI, et al. Common presentations of amebic liver abscess. Ann Emerg Med 1999;34: 351-355.
45. Shandera WX, Bollam P, Hashmey RH, et al. Hepatic amebiasis among patients in a public teaching hospital. South Med J 1998;91:829-836.
46. Nattakom S, Serrato P, Bright T, et al. Amebic liver abscesses masquerading as pyemic abscesses. Clin Infect Dis 2001;33: E145-E147.
47. Seeto RK, Rocky DC. Amebic liver abscess: Epidemiology, clinical features, and outcome. West J Med 1999;170: 104-109.
48. Hughes M, Petri W. Amebic liver abscess. Inf Dis Clin North Am 2000;14:565-581.
49. Panosian C. Parasitic diarrhea. Emerg Med Clin North Am 1991;9:337-347.
50. Miller R, Jong E. In: Jong E, ed. Amebiasis, Giardiasis, and Other Intestinal Protozoal Infections. The Travel and Tropical Medicine Journal. Philadelphia; W.B. Saunders Co.; 1987:325-340.
51. Abdelouafi A, Ousehal A, Ouizidane L, et al. Ultrasonography in the diagnosis of liver abscesses. Annals of Radiology 1993;36:286-292.
52. Kimura K, Stoopen M, Reeder MM, et al. Amebiasis: Modern diagnostic imaging with pathological and clinical correlation. Semin Roentgenol 1997;32:250-275.
53. Ralls PW, Colletti PM, Halls JM. Imaging in hepatic amebic abscess. Amebiasis: Human Infection by Entamoeba Histolytica. New York, John Wiley & Sons; 1988:675.
54. Stanley SL, Jackson TF, Foster L, et al. Longitudinal study of the antibody response to recombinant entamoeba histolytica antigens in patients with amebic liver abscess. Am J Trop Med Hyg 1998;58:414-416.
55. Ravdin JI, Jackson TF, Petri WA, et al. Association of serum antibodies to adherence lectin with invasive amebiasis and asymptomatic infection with pathogenic entamoeba histolytica. J Infec Dis 1990;162:768-772.
56. Rajak CL, Gupta S, Jain S, et al. Percutaneous treatment of liver abscesses. AJR Am J Roentgenol 1998;170:1035-1039.
57. Tandon A, Jain AK, Dixit VK, et al. Needle aspiration in large amoebic liver abscess. Trop Gastroenterol 1997;18: 19-22.
58. Kirchhoff LV. Chagas’ disease. American trypanosomiasis. Infect Dis Clin North Am 1993;7:487-502.
59. Laranja FS, Dias E, Nobrega D, et al. Chagas’ disease: A clinical, epidemiologic, and pathologic study. Circulation 1956;14:1035-1060.
60. Anonymous. Chagas’ disease-interruption of transmission, Brazil. Wkly Epidemiol Rec 1997;72:1-8.
61. Bergoglio RM. Biology of Trypanosoma cruzi. Annu Rev Microbiol 1973;27:347-382.
62. Kirchhoff LV. American trypanosomiasis (Chagas’ disease)—A tropical disease now in the United States. N Engl J Med 1993;329:639-644.
63. Kirchhoff LV, Neva FA. Chagas’ disease in Latin American immigrants. JAMA 1985;254:3058-3060.
64. Maguire JH, Hoff R, Sherlock I, et al. Cardiac morbidity and mortality due to Chagas’ disease: Prospective electrocardiographic study of a Brazilian community. Circulation 1987; 75:1140-1145.
65. Tanowitz HB, Kirchhoff LV, Simon D, et al. Chagas’ disease. Clin Microbiol Rev 1992;5:400-419.
66. Kobayasi S, Mendes EF, Rodrigues MAM, et al. Toxic dilatation of the colon in Chagas’ disease. Br J Surg 1992;79: 1202-1203.
67. Henchal E, Putnak JR. The dengue virus. Clin Microbiol Rev 1990;3:376-396.
68. Anonymous. Dengue/dengue hemorrhage fever: Situation in 2000. Wkly Epidemiol Rec 2000;75:193-200.
69. Da Fonseca L, Antonio B, Fonseca SNS. Dengue virus infections. Curr Opin Pediatrics 2002;14:67-71.
70. Anonymous. Underdiagnosis of dengue-Laredo, Texas, 1999. JAMA 2001;285:877
71. Anonymous. Imported dengue-United States, 1997 and 1998. JAMA 2000;283:1953-1954.
72. Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev 1988;11:480.
73. Isturiz RE, Gubler DJ, del Castillo JB. Emerging and re-emerging diseases in Latin America: Dengue and dengue hemorrhagic fever in Latin America and the Caribbean. Inf Dis Clin North Am 2000;14:121-140.
74. Halstead SB. Dengue hemorrhagic fever. A public health problem and a field for research. Bull World Health Organ 1980;58:1.
75. Halstead SB. The pathogenesis of dengue. Am J Epidemiol 1981;114:632.
76. Anonymous. General considerations. In: Dengue Hemorrhagic Fever: Diagnosis, Treatment, Prevention, and Control, 2nd ed. Geneva: World Health Organization; 1997:1-11.
77. Glader B. and Look K. Hematologic disorders in children from Southeast Asia. Ped Clin North Am 1996;143:665-681.
78. Hurst D, Tittle B, Kleman KM et al. Anemia and hemoglobinopathies in Southeast Asian refugee children. J Pediatrics 1983;102:692-697.
79. Monzon C, Fairbanks V, Burgert E, et al. Hematologic genetic disorders among Southeast Asian refugees. Am J Hematol 1985;19:27-36.
80. Schrier S. Pathobiology of thalassemic erythrocytes. Curr Opin Hem 1997;4:75-79.
81. Weatherall D. Single gene disorders or complex traits: lessons from the thalassemias and other monogenic diseases. BMJ 2000;321:1117-1120.
82. Pearson HA, Cohen AR, Giardina PJ, et al. The changing profile of homozygous b-thalassemia: Demography, ethnicity, and age distribution of current North American patients and changes in two decades. Pediatrics 1996;97:352-356.
83. Schwartz E, Benz E, Forget B. Thalassemia Syndromes. In: Hematology: Basic Principles and Practice, 2nd ed. New York; Churchill Livingston; 1995:486-512.
84. Giardini C. Treatment of b-Thalassemia. Curr Opin Hem 1997;4:79-87.
85. Olivieri N, Brittenham G. Iron-chelating therapy and the treatment of thalassemia. Blood 1997;89:739-757.
86. Bastian H. Hematologic disorders including sickle-cell syndromes, hemophilia, and b-thalassemia. Curr Opin Rheum 1995;7:70-72.
87. Bayless P. Red blood cell disorders. Emerg Med Clinics North Am 1993;11:484-485.
88. Higgs DR. a-Thalassemia. In: Higgs DR, Weatherall DJ, eds. Balliere’s Clinical Hematology. London, WB Saunders; 1993:117-150.
89. Jong E. Blood flukes, liver flukes, lung flukes, and intestinal flukes. In: Jong E, ed. The Travel and Tropical Medicine Journal. Philadelphia; W.B. Saunders Co;1987:474-482.
90. Chin J. Control of Communicable Diseases Manual. Baltimore; United Book Press, Inc.; 2000:115-116.
91. Plourde P, Keystone J. Liver fluke infection. In: Infectious Diseases. Philadelphia; J.B. Lippincott Co.;1994:860-863.
92. Carpenter H. Bacterial and parasitic cholangiits. Mayo Clin Proc 1998;73:473-478.
93. Okolo P, Parsons W. Recurrent pyogenic cholangitis. Emedicine.com. www.emedicine.com;Feb. 11, 2002: vol. 3. Accessed 2-14-2003.
94. Kim TY, Kang SY, Park SH, et al. Cystatin capture enzyme-linked immunosorbent assay for serodiagnosis of human Clonorchiasis and profile of captured antigenic protein of Clonorchis sinensis. Clin Diagn Lab Immunol 2001;8: 1076-1080.
95. Liu L, Harinatsuta K. Liver and intestinal flukes. Gastroenterol Clin North Am 1996;25:627-636.
96. Ross AG, Bartley PB, Sleigh AC, et al. Schistosomiasis. N Engl J Med. 2002;346:1212-1220.
97. Mahmoud A. Tropical Medicine. Infect Dis Clin North Am 1995;9:269-274.
98. Bica I, Hamer D, Stadecker M. Hepatic Schistosomiasis. Infect Dis Clin North Am 2000;4:583-604.
99. de Jesus AR, Silva A, Santana LB, et al. Clinical and Immunologic Evaluation of 31 Patients with Acute Schistosomiasis mansoni. J Infect Dis 2002;185:98-105.
100. Schutte CHJ, Pienaar R, Becker PJ, et al. Observations on the techniques used in the qualitative and quantitative diagnosis of schistosomiasis. Ann Trop Med Parasit 1994;88:305-309.
101. Deelder AM, Qian ZL, Kremsner PG, et al. Quantitative diagnosis of schistosoma infections by measurement of circulating antigens in serum and urine. Trop Geogr Med 1994; 46:233.
102. Tsan VCW, Wilkins PP. Immunodiagnosis of schistosomiasis: Screen with FAST-ELISA and confirm with immunoblot. Clin Lab Med 1991;11:1029-1032.
103. Bosch X. Female genital mutilation in developed countries. Lancet 2001;358:1177-1179.
104. Council on Scientific Affairs, American Medical Association. Female genital mutilation. JAMA 1995;274:1714-1716.
105. Black J, Debelle GD. Female genital mutilation in Britain. BMJ 1995;310:1590-1591.
106. Ford N. Tackling female genital cutting in Somalia. Lancet 2001;358:1179.
107. Wiens J. Female circumcision if curbed in Egypt. BMJ 1997; 314:1148.
108. Fourcroy J. Female circumcision. Am Fam Physician 1999;60:657-658.
109. Committee on Bioethics, American Academy of Pediatrics. Female genital mutilation. Pediatrics 1998;102:153-156.
110. Toubia N. Female circumcision as a public health issue. N Engl J Med 1994;331:712-716.
111. Kandela P. Court ruling means that Egypt embraces female circumcision again. Lancet 1997;350:41.
112. Walder R. Why the problem continues in Britain. BMJ 1995;310:1593-1594.
113. Gallard C. Female genital mutilation in France. BMJ 1995;310:1592-1593.