Diabetic Emergencies
Diabetic Emergencies
Authors: Sonia Angelo, MD, MS, Instructor, Department of Emergency Medicine, Thomas Jefferson Hospital, Philadelphia, PA; and R. Corey Waller, MD, MS, Chief Resident, Department of Emergency Medicine, Thomas Jefferson Hospital, Philadelphia, PA
Peer Reviewers: Sandra M. Schneider, MD, FACEP, Professor and Chair, Department of Emergency Medicine, University of Rochester School of Medicine, Rochester, NY; and Catherine Marco, MD, FACEP, Clinical Professor, Medical University of Ohio, Attending Physician, St. Vincent Mercy Medical Center, Toledo, OH.
Diabetes is one of the most common chronic diseases in man, and as seen below, a frequent cause of emergency department (ED) visits. Diabetics now have more treatment options available, better delivery systems for their insulin and greater technology to allow them to self-monitor their glucose levels. We now know that good control of glucose is the best defense against the deadly long-term consequences of diabetesblindness, renal disease, peripheral neuropathy, etc. The ability to monitor the effects of diabetic control through the measurement of Hg Ac is an important tool in the long-term treatment of diabetics. Unfortunately, good control does not guarantee a lifetime without complications.
While treatments for diabetics become better, the disease itself is more common. In part this is due to the aging of the population. But increasingly the presence of metabolic syndrome in older children has led to an increase in diabetes in the population. This means the ED will see more patients with acute emergencies due to diabetes. Every emergency physician needs to be comfortable with all of the emergencies associated with diabetes. Failure to treat properly carries tremendous consequences, including cognitive impairment or death.
The Editor
Introduction
On a typical day in the ED, it is not unusual to see two to three people during a shift with complaints concerning diabetes. Some of these patients present critically ill and require rapid evaluation and intervention by ED staff. This review discusses the basic physiology of normal glucose regulation, the most common diabetic emergencies, and the prevailing thoughts on appropriate ED intervention.
The highest mortality rates for common diabetic emergencies are for diabetic ketoacidosis (DKA) and hyperglycemic hyperosmolar state (HHS), with mortality rates of approximately 5% and 15% respectively. DKA appears in 4-9% of all hospital discharge summaries, averages $13,000 per visit, and costs more that $1 billion per year nationwide.1-4 For HHS, the figure is more difficult to derive due to the lack of population-based studies on the topic, but some estimates make it fewer than 1% of all diabetic admissions.5
Hypoglycemia, while less costly, has admission rates as high as 4.7 per 1000 person-years (PY) and as high as 7.1% per year in insulin-dependent diabetics and 7.3% per year in non-insulin dependent diabetics.6,7 This most likely is an under-estimation since as many as one-third of these patients are treated by prehospital personnel and never transported to the hospital. In addition, countless patients also self-treat and never report the incident. Hypoglycemia is the most common complication in people taking oral hypoglycemic medications. A large proportion of the diabetic population self-treats at home without ever involving either prehospital or hospital personnel.
Normal Physiology
Insulin is a hormone secreted by the cells of the pancreas in reaction to an increase in glucose concentration, as well as many other substances such as amino acids, free fatty acids, gastrointestinal (GI) hormones, and sympathetic output. When insulin is released from the pancreas, it has both immediate and long-term effects on the body's storage and utilization of glucose.
When glucose is ingested, it enters the GLUT 2 channel on the cell, closes ATP dependent K+ channels, and opens voltage gated Ca++ channels causing an influx of calcium, which attaches to and activates motor proteins releasing the insulin. (See Figure 1.) While this is a simplified version of the actual mechanism, the basic premise is repeated with any carbohydrate load.
| Figure 1. Mechanism for Insulin Release from Beta Cell |
|
|
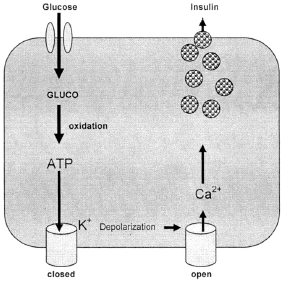
When insulin is released, it binds to a cell's insulin receptor and turns on the enzyme-linked machinery. The insulin receptor works through a tyrosine kinase signaling pathway, which acts by phosphorylating multiple intracellular enzymes of the insulin-receptor substrate class (IRS). Different IRS enzymes are located in each tissue type and have moderately different effects depending on the tissue.8,9 (See Figure 2.)
| Figure 2. Insulin Receptor Signaling Pathway |
|
|
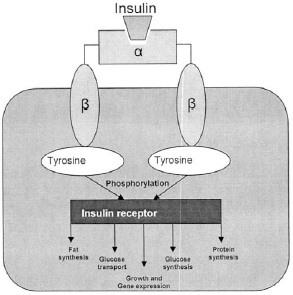
Within seconds of insulin binding to the receptors, the membranes of about 80% of the body's cells dramatically increase their glucose uptake. This happens more in muscle and adipose cells and less in neurons. Neurons are not dependent on insulin for glucose uptake and thus rely more on the concentration gradient imposed by glucose load. This subsequently plays a role in cerebral edema seen with very high glucose levels. Once insulin is no longer available, the glucose transporters are adsorbed into the cell for use later. Soon after this, the cell membrane has increased permeability to amino acids, potassium ions, and phosphate ions.
Slower effects include the change of intracellular glucose storage enzymes, and an increase in the activity of mitochondria and many tissue-specific mechanisms. This happens over the course of about 10-15 minutes. Then, over the next few hours and even days, the rates of mRNA translation and DNA transcription increase.10,11
Two tissues of interest are muscle cells (non-cardiac) and adipocytes. In muscle, glucose is stored as glycogen at a concentration of up to 3% in the cytosol. This can be used for short periods of extreme energy requirements and during anaerobic exercise. No matter what the oral intake, a lack of insulin can result in decreased energy stores and, thus, decreased exercise tolerance. The presence of insulin in adipose tissue inhibits the action of hormone-sensitive lipase. This enzyme is responsible for hydrolysis of triglycerides stored as fat, which inhibits fatty acid release into the blood. When glucose is transported into the adipocyte, the majority of it is used to synthesize alpha-glycerol phosphate, which is the backbone for fatty acids. The low insulin levels essentially block the storage of glucose as fat. While this sounds helpful, it leads to an abnormal increase in the circulating fatty acids. This happens in two ways: 1) the action of hormone sensitive lipase is increased, releasing large amounts of fatty acids from stored fat; and 2) lipoprotein from the liver is blocked from storage in adipocytes. In the absence of insulin, the free fatty acid concentration rises almost immediately.12 This is the first step in the formation of the keto-acids responsible for the acidosis in DKA.
Glucagon is a hormone secreted by the cells of the pancreas and acts to reverse and/or mitigate the effects of insulin on the body. Less is known about the cellular signaling of glucagons, but, in general, low glucose levels will increase the secretion and high amino acid levels will induce secretion. Once glucagon is secreted, liver glycogen is broken down to release glucose, and gluconeogenesis is ramped up. A dose of 1 mcg/kg of glucagon can increase the blood glucose by as much as 20 mg/dL in about 20 minutes.13
Classifications and Diagnostic Criteria
In general, the diagnosis of diabetes mellitus involves a fasting glucose greater than 126 mg/dL (7.0 mmol/L) or a random glucose of greater than 200 mg/dL (11.1 mmol/L) with the middle ground of "impaired fasting glucose" being a glucose concentration between 100 and 125 mg/dL (5.6 - 6.9 mmol/L). However, the reason for this perturbation in carbohydrate metabolism is what drives both the treatments and probable complications of the disease. Therefore, it is necessary to further classify the disease based on the cause.14
Diabetes terminology has changed during the past 10 years to accommodate the ever-increasing specificity with which the disease is diagnosed. In the past, diabetes was classified as juvenile-onset, insulin-dependent, non-insulin-dependent, and/or adult-onset diabetes. These classifications have given way to the terms type 1 and type 2 diabetes.15
Type 1 diabetes involves the destruction of pancreatic cells that leads to an absolute insulin deficiency. This can be determined by the presence of autoimmune antibodies such as islet-cell antibodies (ICA), glutamic acid dehydrogenase (anti-GAD), or anti-insulin antibodies. Those patients who have no evidence of the antibodies but destruction of cells on pancreatic biopsy are diagnosed with idiopathic type 1B diabetes mellitus.16 Patients who are diagnosed with type 1 diabetes have an absolute insulin requirement and can quickly develop DKA in its absence.17
The diagnosis of type 2 diabetes is less absolute because no specific diagnostic test exists. Patients with type 2 diabetes have varying amounts of insulin deficiency and resistance. Patients with type 2 diabetes will have hyperglycemia and possibly hyperosmolar hyperglycemic coma, but typically are not in pure DKA. Recent studies demonstrate that obese black patients who have been diagnosed with type 2 diabetes have an increased risk of developing DKA. These patients typically are not found to have any antibodies seen in type 1 diabetics, but rather have an overt resistance to insulin so that lipid metabolism is shunted toward the production of ketoacids.18,19 Because of the inconsistencies with this small population of diabetics, a third group of patients may fit into an atypical disease category.20 All of the above categories have associated complications, including DKA, HHS, and hypoglycemia.
The diagnosis of DKA is made up of a triad of laboratory findings that include hyperglycemia, ketonemia, and acidosis. Independently, each of these can be caused by many different aberrations, but together signal a downward spiral of dehydration and poor perfusion. Attempts have been made to classify DKA into mild, moderate, and severe categories (see Table 1) which, in turn, dictate the level of aggression used in treatment.21 Mild, moderate, and severe DKA are differentiated based on the level of acidosis (the pH), the depletion of the buffer system (HCO3), and the level of consciousness. Patients in mild DKA have an arterial pH of 7.25-7.30, a serum bicarbonate of 15-18 mEq/l, and a normal sensoria. Patients with moderate DKA have an arterial pH of 7.0-7.24, a serum bicarbonate of 10-15 mEq/l, and are either alert or drowsy. Severe DKA patients have a pH of less than 7.0, a bicarbonate of less than 10 mEq/l, and they are either stuporous or are in a frank coma.
| Table 1. Diagnostic Criteria for DKA and HHS |
|
|

HHS, which has gone by other names such as hyperglycemic hyperosmolar nonketotic state and hyperosmolar nonketotic coma, now holds the more general title, leaving out the designation of "nonketotic." This is due to the fact that small amounts of ketones may be present and a change in sensorium may occur at many levels including, but not limited to, coma. Generally patients with HHS have a serum glucose greater than 600 mg/dL, a pH greater than 7.3, a serum bicarbonate greater than 15 mEg/l, small or no ketones, and a serum osmolarity greater than 320 mOsmol/kg. The mental status changes are due to the hyperosmolar state and may range from mild confusion to unresponsiveness.
Hypoglycemia is defined as a blood sugar below that of the normal range of 50 mg/dL (3.8 mmol/l) and may be asymptomatic in someone with poor glycemic control. For the purposes of this review, hypoglycemia will be defined as a diabetic patient who requires intervention with glucose or glucagon.
Pathophysiology
DKA and HHS share many common pathways and may occur as an overlapping state. However, the biochemical pathway of DKA is better understood than that of HHS. As discussed earlier, the primary perturbation is a decrease in the physiological doses of insulin needed (or increased insulin resistance) to maintain homeostasis of the glycolytic pathway, or an increase of stress hormones that cause a relative decrease in insulin. This can occur during infection or trauma by causing a baseline insulin resistance to increase glucose concentrations to the level necessary for the increased ATP requirement of damaged or over-worked cells. There also is a concomitant increase in glycogenolysis secondary to increased catecholamines, which releases stored glucose into the system.
When this occurs, hyperglycemia begins the downward spiral of dehydration secondary to osmotic diuresis; decreased end-organ perfusion; further increases in the stress hormones epinephrine, glucagon, cortisol, and growth hormone; and then the beta-oxidation of fatty acids, which produce ketone bodies that, in turn, cause an acidosis. (See Figure 3.) This last part is where DKA and HHS differ in their pathophysiology. In HHS there seems to be enough baseline insulin to quell the breakdown of fatty acids, thus keeping the ketone bodies to a minimum. There also seems to be a decrease in the amount of growth hormone in the plasma; however, there have not been any definitive studies that show how this impacts free fatty acid release. It is postulated that the severe dehydration and impaired renal dysfunction seen more in HHS are secondary to the gradual onset as well as the inability of glucose to be excreted through the urine at the same rate as in DKA. There also is less of a polydipsic response associated with HHS than with DKA, contributing to the relatively higher glucose concentrations. This decrease in oral intake possibly is worsened by the change in mental status seen in patients with HHS.
Electrolyte disturbances are a common aspect of DKA and HHS that require strict monitoring and correcting along with the hyperglycemia and acidosis. The major electrolytic disturbances are with sodium and potassium. Both are excreted along with anions during the osmotic diuresis in DKA and HHS, and also are shifted into different compartments with the acidosis and dehydration. As the proton concentration in the intravascular volume rises, the H+/K+ exchanger increases its rate of exchange, attempting to equilibrate the pH. This artificially drives the serum [K+] up and can lead to a false sense of security on a chemistry panel. Sodium may be falsely lowered due to the dilutional effect of a high glucose and must be corrected.
The hypoglycemic syndrome can include CNS dysfunction such as sweating, tachycardia, confusion, tremor, extremely bizarre behavior, stroke-like symptoms, and overt coma. The early symptoms arising from hypoglycemia are caused by the release of epinephrine. All patients with neuroglycopenic effects like mental status changes should have a bedside capillary glucose test as soon as possible. Unfortunately, those patients who have experienced hypoglycemia in the past may not show these signs early in the process, which can result in profoundly low glucose levels for extended periods of time. The pathogenesis of hypoglycemia in diabetics is rather straightforward when insulin is the drug, but can be more problematic in situations such as medication overdose and hypoglycemia in renal failure or sepsis.
Hypoglycemia secondary to diabetic medication overdose is much more common than hypoglycemia caused by renal failure or sepsis and most overdoses are unintentional, occurring secondary to dementia or in patients who forget to eat in time. However, some instances are purposeful and, given a suspicious situation, the physician should investigate further. Depending on the half-life of the insulin or oral hypoglycemic agent, these patients will have high glucagon and catecholamine levels leading to the possibility of rebound hyperglycemia after self-treatment. In renal failure, the hypoglycemia arises from decreased clearance of either the insulin or hypoglycemic medication. The most common drugs other than insulin that causes hypoglycemia are the sulfonylureas, some of which have extended pharmacological half-lives.
In the nondiabetic patient who presents with hypoglycemia conditions such as reactive hypoglycemia, liver disease, Addison's disease, insulin-producing tumors, alcohol ingestion, and factitious hypoglycemia should be considered.
Hypoglycemia from alcohol ingestion typically results from lack of substantive oral intake and a decrease in gluconeogenesis after alcohol ingestion.22 Factitious hypoglycemia can result from injection of insulin or ingestion of sulfonylureas. In the case of insulin injection, a serum c-peptide and insulin level can be drawn. If the ratio of insulin to c-peptide is greater than 1, then self-injection is likely. If it is lower than 1, then other causes, including an insulin-secreting tumor, should be sought.
Common Diabetes Treatments and their Side Effects
Insulin is the oldest and most important treatment for type 1 diabetes. It is available in rapid-, short-, intermediate-, and long-acting forms that may be injected separately or mixed in the same syringe. In the past, physicians only had to be concerned about an overdose of regular insulin, which, when injected SQ, has a peak effect in about one hour and wears off by approximately 4 hours. Today, however, there are many types of insulin that are produced from recombinant human forms as well as swine derivatives.
There are four general types of insulin available. Rapid-acting insulin analogs (Lispro and Aspart) act in 5-15 minutes and are used in insulin pumps. Regular insulin (onset of action 30-60 minutes when given SQ) still is very common and is considered a short-acting insulin. Intermediate-acting Lente and NPH insulin start working within 2-4 hours after injection and can last as long as 20 hours. Insulin preparations with a predetermined proportion of NPH mixed with regular, such as 70% NPH to 30% regular, are considered intermediate-acting but have a short-acting component. The only long-acting insulin is Ultra-Lente and its duration of action is 18-24 hours. (See Table 2.) Human insulin has a more rapid onset and shorter duration of activity than swine-derived insulin.
| Table 2. Types and Actions of Insulin* |
|
|

Insulin is commercially available in concentrations of 100 or 500 U/mL (designated U-100 and U-500; 1 U equals ~ 36 mcg of insulin). The U-500 concentration is used only in rare cases of insulin resistance when the patient requires extremely large doses. The U-500 and the insulin analog Lispro are the only insulins that require a prescription. Insulin preparations sometimes are formulated individually for use in infants (e.g., U-10) with diluents provided by the manufacturer. In these instances, special care must be taken to ensure that the correct dose of the diluted insulin is administered with an ordinary insulin syringe.
Different types and species of insulin have different pharmacological properties. Human insulin is preferred for use in pregnant women, women considering pregnancy, individuals with allergies or immune resistance to animal-derived insulin, those initiating insulin therapy, and those expected to use insulin only intermittently. Insulin type and species, injection technique, insulin antibodies, site of injection, and individual patient response differences can all affect the onset, degree, and duration of insulin activity. Changing insulin types may affect blood glucose control.
Given all of the above information, it is not surprising that many patients accidentally overdose on their insulin. It also is worth noting that regular insulin can be purchased without a prescription. This is important since it is a readily available drug that can be used for overdose and/or factitious hypoglycemia.
Oral medications to control both type 1 and type 2 diabetes are extremely common and freely prescribed. Oral antidiabetic agents can be grouped best by their treatment function. This includes medications that decrease the rate of carbohydrate breakdown or absorption, agents with a mechanism of action that improves endogenous insulin sensitivity (i.e., biguanides, thiazolidinediones, or combinations of these), as well as agents that are considered endogenous insulin secretagogues (i.e., sulfonylureas and meglitinides). (See Table 3.)
Alpha-glucosidase inhibitors lower blood glucose by modifying the intestinal absorption of carbohydrates and fat. The most common of these is acarbose, which has only minor side effects, including bloating, diarrhea, and flatulence.23 These have not been shown to increase the risk of hypoglycemia.
There are two classes of hypoglycemic drugs that act by improving insulin action on cells. These are thiazolidinediones and biguanides. The thiazolidinediones (glitazones) include rosiglitazone (Avandia) and pioglitazone (Actose), while the biguanide group includes only metformin.
Thiazolidinediones work by modulating insulin sensitivity through the peroxisome proliferators-activated receptor complex (PPARs) located in adipose tissue, pancreatic cells, vascular endothelium, and activated macrophages.24-26 Ultimately, the exact mechanism is not known, but the effect is both an increase in glucose utilization and decrease in glucose production by the liver and muscle cells. Possible issues with these drugs are hepatotoxicity,27,28 weight gain, and, most important for the emergency physician, congestive heart failure (CHF).29,30 CHF is thought to be secondary to the alteration of action the PPARs have on the nephron. Sodium reabsorption by the collecting tubule cells in increased which, in turn, increases total circulating volume.31
Metformin is prescribed more commonly than the glitazones and causes the same net effect of increased insulin sensitivity. The mechanism by which metformin achieves this is thought to be via activation of AMP-activated protein kinase; however, the pathway has not been worked out completely.32-34 While metformin has been shown to greatly enhance the control of type 2 diabetes and even to lower lipids, it does have two major associated complications. The first, and most common, is abdominal discomfort and nausea. This complication also can be associated with diarrhea and a persistent metallic taste.33 The second and most devastating is lactic acidosis. Due in part to a change in prescribing habits (i.e., not prescribing it to those with known predisposing factors), this now is a rare but very real potential side effect. Predisposing factors for the development of lactic acidosis include renal failure (serum Cr above 1.4 mg/dL in women and 1.5mg/dL in men), liver disease or heavy alcohol use, CHF, past history of lactic acidosis, and acute illness.35 The treatment of this condition will be discussed in the treatment section of this review.
Another major class of oral diabetes medications is the sulfonylureas. These are the most widely used drugs in the treatment of type 2 diabetes.36 This class includes the short-acting medications glipizide (Glucotrol, Glucotrol XL) and tolazamide (Tolinase), and the long-acting versions chlorpropamide (Diabinese), glimepiride (Amaryl), and glyburide (DiaBeta, Micronase, Glynase). Both the long- and short-acting drugs work by binding to the sulfonylurea receptor, which is a component of the ATP-dependent potassium channel in the pancreatic beta-cell.37 The activation of this channel then opens the voltage gated calcium channel, leading to calcium influx, fusion of insulin-containing vesicles to the internal membrane, and insulin release. Overall, this leads to a more vigorous release of insulin when the glucose level rises.
The meglitinides (Repaglinide and Nateglinide) are another class of oral hypoglycemic drugs that, while working on a different receptor than the sulfonylureas, yield the same result. Both of the available drugs are shorter-acting and are taken before meals, are metabolized by the liver, and are excreted renally. In the setting of renal insufficiency, Nateglinide should be used cautiously since, unlike Repaglinide, it has active metabolites that are excreted renally.
Sulfonylureas and meglitinides represent the oral diabetes agents with the greatest potential for causing hypoglycemia. Hypoglycemia is more likely to occur in the setting of exercise after missing a meal, recent increase of the drug, use of the longer-acting drugs, and alcohol use or abuse. In the setting of other drugs such as warfarin, salicylates, sulfonamides, or fibric acid derivatives, the level of hypoglycemic agents may be increased or have a prolonged circulatory time.38 In overdoses of these medications, hypoglycemia may be difficult to control and may warrant an intensive care unit (ICU) or step-down unit admission.
Precipitating Factors for Hyper- and Hypoglycemic Complications
Traditional thoughts of DKA exacerbation lean toward the presence of an infection, such as pneumonia or urinary tract infection, as the cause of insulin and glucagon disturbances. However, in certain populations it is lack of insulin, either by omission or underdosing, that may be the sole precipitating factor for the hyperglycemia and release of other hormonal modulators that induce DKA. This has been seen particularly in the African American population, who may present in DKA without a previous diagnosis. In 20% of cases, patients may be in the ED without prior diagnosis of diabetes, and therefore may not have received any intervention.21
Infections do instigate 4-36% of DKA episodes, with pneumonia and urinary tract infections accounting for 30-50% of these.21 Other precipitating factors include alcohol abuse, trauma, myocardial infarction, and pulmonary embolism, all occurring in both insulin-dependent diabetes mellitus and non-insulin-dependent diabetes mellitus. Drugs such as corticosteroids, beta-blockers, diuretics, and sympathomimetics also can instigate episodes of DKA. An even newer risk for DKA is the insulin pumps, which in the mid-1990s were shown to double the risk of DKA when compared to multiple daily injection groups.3 This was thought to be an effect of the ultra-short-acting insulin used in these pumps and undetected malfunction. More recent studies, however, have shown that they are superior to conventional methods of treatment.39
Other reasons for increased risk of DKA include noncompliance, eating disorders (up to 20% of recurrent DKA in young adults), and lack of adequate housing and/or health insurance. The socioeconomic aspects of this disease are numerous but are outside the scope of this review.
Diagnosis
As with all ED patients, physicians must take care to assess the airway, breathing, circulation, and mental status of patients with potential diabetes-related emergencies. Once the basics are accomplished, then assessment of renal function, hydration status, and the search for sources of infection should follow. A history of polyurea, polydipsia, weight loss, nausea, and vomiting are vital to the narrowing of the differential. A sweet odor on the breath, loss of skin turgor, dry mucous membranes, tachycardia, and hypotension can be found in patients with moderate to severe DKA and HHS (no sweet odor on the breath for HHS). It should be noted that not all people can detect the sweet odor associated with the presence of ketones. One of the hallmarks of HHS is an altered mental status, which can encompass anything from mild confusion to overt coma. Three rapid bedside laboratory studies that can be preformed are the determination of capillary glucose, urine for specific gravity, and the presence of glucose, ketones, and, in some cases, serum acetone.
Aside from the usual first-line laboratory values of a complete blood count (CBC), Chemistry 7, and blood cultures, arterial/venous pH should be taken, serum osmolarity measured, and ketones quantified. A venous pH is as accurate as an arterial blood gas if 0.03 is added to the pH and 1.2 mmol/L is added to bicarbonate.40,41 This may be helpful in quick assessment of hypotensive or pediatric patients.
Ketones should be measured and followed throughout the treatment to assess for adequate cessation of ketogenesis. The ketone of choice should be beta-hydroxybutyrate rather than acetone, simply because acetone can drastically underestimate the ketosis present. This occurs because beta-hydroxybutyrate production from acetoacetate is the preferred by-product rather than acetone.42,43 (See Figure 3.) If the beta-hydroxybutyrate level is not available, hydrogen peroxide may be added to the urine before testing for acetone. This will convert the beta-hydroxybutyrate to acetoacetate and increase the chance for a positive confirmatory test. Remember that patients with HHS still may have a mild ketosis. It is important always to measure and calculate the serum osmolarity to search for an unmeasured gap (such as methanol or ethanol), thus making sure that the hyperosmolarity is responsible for a mental status change. If serum osmolarity is less than 320 mOsm/Kg, another source for the mental status change should be sought. Standard calculations for serum chemistry values are outlined in Table 4.
| Table 4. Calculations Using Serum Chemistries |
|
|

Treatment
The treatment of DKA and HHS must include the correction of the three derangements of hyperglycemia, dehydration, and electrolyte deficiencies. Guidelines for the treatment have been delineated by the American Diabetes Association and are shown in Figure 4.
The theory and practice revolve around the rapid rehydration of the patient, which increases renal perfusion and helps eliminate excess glucose, ketone bodies, and fixed acids. Secondly, insulin is given to halt the liberation of free fatty acids and ongoing gluconeogenesis. Lastly, electrolytes lost from the osmotic diuresis are replaced. Each of these three interventions should be done in a controlled manner with close monitoring of glucose, electrolytes, and osmolarity.
Patients with mild DKA (venous pH >7.29, serum HCO3 >16, no neurological impairment, and less than 3% volume depletion) can be managed in the ED and then discharged home with adequate follow-up.21 The above does not pertain to children, adolescents, pregnant women, or renal failure patients. See the section on special populations for appropriate treatment and disposition of these groups. If the patient is not vomiting, use of oral rehydration solutions are sufficient to replenish the mild dehydration. Use of short-acting subcutaneously injected insulin at a dose of 0.4 units/kg is appropriate in the office, but in the ED intravenous insulin is preferred because of its more rapid onset. Finger-stick glucose checks every hour to ensure a proper response are important. Initial electrolytes should be checked for hypokalemia and, if found, oral supplementation may be used, but IV KCl is more predictable (due to changes in GI absorption in DKA) and is preferred. Most often, the condition can be corrected within 4-6 hours. A recheck of electrolytes and venous gas before discharge is necessary to evaluate for completeness of treatment. If follow-up is questionable, then these patients should be admitted for a short time to ensure stability.
Initially, normal saline (0.9% NaCl) is given at a rate of 20 cc/kg or 1-1.5 liters for an adult over the first hour. After the initial rehydration, a bedside glucose may be done to assess the level of hyperglycemia remaining.
In moderate or severe cases of DKA, there may be a rapid change in glucose that brings the level close to normal so that insulin therapy may be needed only for a short time. Electrolytes also should be checked and remaining fluid therapy can be changed based on the corrected serum sodium level. If the corrected sodium level in a patient with DKA is normal or increased, then half normal saline (0.45% NaCl) may be used and given at a rate of 4-14 mL/kg/hr. If the level is low, then 0.9% NaCl may be given at a similar rate. if hypokalemia is present initially, IV KCl replacement is indicated, as correction of any acidosis will make the serum hypokalemia worse. The replacement of the fluid deficit should be done over a period of 48 hours. The serum osmolarity should be followed and not be decreased more than 3 mOsm/kg/hr. If corrected any faster, iatrogenic cerebral edema may result from the flow of free water down the concentration gradient into the neuronal tissues, causing mental status changes and permanent damage. This mainly is a concern in the pediatric population, although it still can be an issue in adults with HHS. Patients also should be monitored for signs of fluid overload such as CHF. Strict intake and urine output should be monitored to maintain a net positive in fluid rehydration.
Treatment of HHS follows the same principles as are given for moderate to severe DKA. However, patients with HHS have more severe fluid deficits and are less likely to have problems with potassium. Many are not known to be diabetic and may never have been given insulin before. Smaller doses of insulin initially, especially during aggressive fluid replacement, will help to prevent hypoglycemia.
Insulin therapy should be used for all cases of DKA and HHS. In known diabetics with mild DKA, subcutaneous/IV administration of 0.4-0.6 units/kg of regular insulin may be sufficient with capillary glucose checked and covered with a standard sliding scale if/when admitted to the floor for monitoring. If the patient is newly presenting with diabetes and in DKA or HHS, a lower dose of regular insulin may be used (0.1-0.15 U/kg) secondary to the probable increase in sensitivity to insulin. In moderate to severe cases of DKA, (venous pH < 7.25, serum HCO3 <10, severe to no neurological impairment, and greater than 10% volume depletion), IV bolus followed by continuous IV administration of regular insulin is the standard. Once hypokalemia has been excluded, an intravenous bolus of 0.15 units/kg of body weight should be given and followed by a regular insulin drip at 0.1 units/kg/hr (5-7 units/hr). A steady decline of serum glucose by 50-75 mg/hr is the goal of the continuous infusion, and adjustment of rate may be necessary. If hypokalemia is present, then IV KCl should be given at a rate not to exceed 20 mEq/hr, with addition of 40 mEqs of oral KCl (if patient can tolerate PO). Continue rehydration until K+ is above 3.3 mEq/L, then institute insulin therapy as outlined above.
The replacement of electrolytes, specifically potassium, is of the utmost importance in the maintenance of good cardiac function. To prevent continued or worsening hypokalemia, potassium replacement should begin when its level goes below 5.5 mEq/L in the setting of adequate urine output. If potassium concentration is less than 3.3 mEq/L, replacement should begin immediately and precede any administration of insulin. Typically, 20-30 mEq/L (two-thirds KCl and one-third KPO4) in each liter of fluid after the initial bolus therapy is adequate to maintain a normal potassium level.
Once the plasma glucose level reaches 250 mg/dL in DKA and 300 mg/dL in HHS, the insulin drip may be decreased to 0.05-0.1 units/hr. The IV fluid should be changed to a dextrose-containing solution such as D5 0.45 NaCl if the corrected sodium is high or normal, and D5 0.9% NaCl if the corrected sodium is low. Insulin therapy should be continued for as long as ketones are present in the serum for DKA or until electrolytes and mental status is at baseline for HHS.
The need for bicarbonate in DKA has been debated over many years and only a general consensus exists. When a patient's pH is greater than 7.0, then no HCO3 is needed. If the pH is between 6.9 and 7.0, 50 mmol in 200 cc of water over 1 hour should be given. If the pH is less than 6.9, 100 mmol in 400 cc of water should be infused over 2 hours. Since bicarbonate also decreases potassium, care should be taken to closely monitor its serum level.
Resolution of DKA includes a combination of a serum glucose less than 200 mg/dL, serum bicarbonate greater than 18 mEq/L, and a venous pH of greater than 7.3. Once this is reached, the IV insulin may be decreased slowly and overlapped with and then replaced by subcutaneous administration as needed every 4 hours with continuation of oral food intake. Resolution of HHS takes place when baseline mental status has been reached (sometimes over days) and plasma glucose is greater than 200 with serum osmolarity in the normal range. Blood should be drawn every 2-3 hours to monitor electrolytes, serum osmolarity, creatine, beta-hydroxybutyrate, and venous pH.
Patients who have a mild case of DKA can be admitted to the medicine floor safely, while those with HHS or moderate to severe DKA should be admitted to either an intermediate care bed or an ICU. Rarely, patients can be corrected in the ED and discharged home. If this is deemed appropriate, then close follow-up is required. If that is not possible, a follow-up visit the next day for a capillary blood glucose check and evaluation of hydration status should be set up.
Hypoglycemia
The treatment of hypoglycemia (blood glucose below 50 mg/dL) is relatively straightforward, but if done incorrectly or hastily can have profound long-term effects. The first determination that should be made is what caused the hypoglycemia. If it is purely secondary to taking the regular dose of insulin and/or oral hypoglycemics but having inadequate PO intake, then 100 cc of 50% dextrose (4 cc/kg 25% dextrose for children) can be given, followed by a regular meal. This has been shown to be adequate as long as the patient can tolerate PO and does not have a current infection. If possible these patients should be monitored in the ED for 1-2 hours. If the patient is at home or does not have IV access, then glucose gel or glucagon at a dose of 1-2 mg to an adult patient (0.5-1 mg in a pediatric patient) may be given. This should be followed by a meal.
Evidence suggests that 15 g of glucose [15 g of glucose in the form of glucose tablets, 15 mL (3 teaspoons) or 3 packets of table sugar dissolved in water, 175 mL (3/4 cup) of juice or regular soft drink, 6 small hard candies such as Life-Savers (1 = 2.5 g of carbohydrate), 15 mL (1 tablespoon) of honey (monosaccharide)] is required to produce an increase in blood glucose of approximately 37.8 mg/dL (2.1 mmol/L) within 20 minutes, with adequate symptom relief for most people. This has not been well studied in patients with gastropathy. A 20-g oral glucose dose will produce a blood glucose increment of approximately 64.8 mg/dL (3.6 mmol/L) at 45 minutes. Other choices such as milk and orange juice are slower to increase blood glucose levels and provide symptom relief. Glucose gel is quite slow [< 18 mg/dL (< 1.0 mmol/L) increase at 20 minutes] and must be swallowed to have a significant effect. (Patients taking an alpha-glucosidase inhibitor (i.e., acarbose [Prandase]) must use glucose (i.e., dextrose) tablets or, if those are unavailable, milk or honey to treat hypoglycemia.) Glucagon 1 mg subcutaneously or intramuscularly produces a significant increase in blood glucose (from 36-216 mg/dL or 3.0-12.0 mmol/L) within 60 minutes.44,45
If the hypoglycemia is due to an overdose of insulin or oral hypoglycemic, then the initial step remains the same, giving 100 cc of 50% dextrose (4 cc/kg 25% dextrose for children), but glucagon may be added for a more prolonged response. One to two milligrams of glucagon should be given via IV bolus (the bolus may be repeated up to three times) followed by a drip of 1 to 2 mg/hour until the blood sugar stays above 100 mg/dL for 3-4 hours. Once this has been achieved, then the glucagon drip may be titrated down and oral glucose therapy should be instituted. If glucagon alone does not maintain the blood sugar above 80 mg/dL, then continuous IV glucose should be started. A drip of 10-20% dextrose may be used and titrated to maintain blood glucose greater than 80-100 mg/dL. This should be done through a central venous catheter to lessen damage to peripheral veins.46
Severe, sustained hypoglycemia from an overdose may require prolonged support. A poison control center may be helpful in assisting with the use of drugs that may improve glucose levels, such as octreotide.
In the case of overdose with a sulfonylurea medication, octreotide may be used to decrease the rebound hypoglycemic effects commonly seen. This is given at a dose of 50-100 mcg orally every 6-12 hours or, if the patient is unable to take PO, then IV administration can be done by diluting the dose in 50 cc of normal saline and infusing it over 15-30 minutes.47
Any patient who overdoses on any oral antihyperglycemics or long-acting insulin should be admitted for close monitoring in a step-down unit or intensive care setting. All other overdose possibilities, such as aspirin, acetaminophen, and elicit drugs should be explored in the ED.
Special Populations
Pediatrics. In pediatric patients, the first presentation of type 1 diabetes is commonly DKA. DKA is the leading cause of morbidity and mortality in children with type 1 diabetes mellitus and, for unknown reasons, is more likely to occur in children with type 2 diabetes than in adults with type 2 diabetes.19,48 The same criteria for diagnosing DKA in adults apply to children, but children may present in profound extremis secondary to lack of continued oral hydration in the setting of persistent osmotic diuresis. This is especially true of infants who are unable to express thirst. In these cases, lethargy, irritability, and weight loss are common signs of DKA in infants. In the early stages of the process, they may not show the normal signs of hypovolemia such as increased pulse or hypotension. Using the anion gap in children is a dangerous way to predict the severity of acidosis or ketosis.49 This is because children tend to have normal renal function and can excrete as much as 30% of their acid load in the urine, as well as depleting sodium and potassium, thus limiting the size of the anion gap. Direct measurement of the beta-hydroxybutyrate is the recommended way to assess baseline and resolution of ketosis.
Fluid repletion should start with 10 cc/kg of isotonic saline or lactated ringers (lactate may be better since it contains potassium and lactate, which can be converted to bicarbonate) over 1 hour. If the patient has circulatory compromise, then this may be repeated a second time. Once the patient is hemodynamically stable, a solution comprised of 0.45% saline with 20 mEq/L of potassium acetate and 20 mEq/L of potassium phosphate and be given at a maximum of 1.5 to 2 times maintenance (provided there is adequate urine output). Faster rates have been associated worsening of the hyperchloremic metabolic acidosis and should be avoided.50,51
Initial insulin should be started lower than the dose for adults at a dose of 0.05 to 0.1 units/kg/hr, given the greater sensitivity of insulin. Once the glucose level is below 250-300 mg/dL (13.9 to 16.7 mmol/L) the IV solution should be changed to D5LR or D5NS. If the glucose is less than 200 mg/dL, then the dextrose may be increased to 7.5 or 10%.49
Children and adults alike should be monitored closely, with electrolytes and pH evaluated hourly for the first four hours. Assessment of the pediatric patient for headache, reduced level of consciousness, bradycardia, and hypertension is important to decrease possible morbidity and mortality from cerebral edema.
Cerebral edema is thought to be present at some level even before the initiation of treatment and then exacerbated by appropriate or inappropriate treatment of DKA. Most of the cases have been noted at 4-12 hours after the initiation of treatment. The following risk factors have been elucidated:52
- children with newly diagnosed diabetes mellitus;53
- younger children;54
- increased BUN and or more profound hypovolemia; and
- the use of bicarbonate therapy.
Several myths about the cause of cerebral edema have recently been shown not to increase risk. These include rate of glucose decrease, rate of insulin delivery, initial glucose and sodium concentrations, and rate of fluid delivery.53,55 Cerebral edema, when present, was thought to be related to the osmotic shift of free water across the blood-brain barrier in reaction to the high concentration of glucose. However, this mechanism alone does not explain why pediatric patients are at greater risk than adults and why not all patients are susceptible. The only consistent perturbation is the presence of insulin therapy. More research is needed to elucidate the actual mechanism.
The Pregnant Patient. Overall pregnant diabetics have a more difficult time maintaining euglycemia. Despite this, the incidence of DKA is relatively low (1-3%) compared to the rest of the population.56 The presentation of a pregnant woman in DKA is not unlike that of the non-pregnant person, with one interesting caveat: the relatively high percentage (up to 36%) who present in DKA with blood glucose levels less than 200 mg/dL.57 With the exception of close fetal monitoring, these patients should be treated the same as non-pregnant patients.
The Renal Failure Patient. Renal failure patients come in two distinct groups: those who still produce urine and are not on dialysis, and those who are on dialysis and are extremely oliguric or completely anuric. For the patient who is dialysis-dependent, DKA does not involve hypovolemia and can be treated by insulin drip (2-3 units/hr) and electrolyte monitoring. If the potassium is greater than 6 mEq/dL, dialysis can be considered. For patients not on dialysis, a less aggressive approach to fluid replacement should be used and a lower starting dose of insulin should be used.58,59 Hypoglycemia is a more common complication with these patients and usually is due to decreased excretion of insulin or oral diabetic medications. This is treated as outlined above for the average patient. Potassium replacement should be done very cautiously in this group secondary to lack of excretion. The mortality rate of this group can be much higher than that of other groups, so admission to a step-down or ICU bed is preferable.
Summary and Pitfalls
In treating diabetic emergencies in the ED, physicians should keep in mind the following summary points and pitfalls:
- Type 1 and type 2 are the preferred designations for diabetes.
- Polyurea, polydipsia, weight loss, and nausea are the most common presenting signs. Lethargy is a late and ominous symptom of DKA.
- The cornerstones of the treatment of DKA and HHS are fluid replacement and insulin. Both should be started early in the ED. Do NOT wait until the patient is admitted to start an insulin drip.
- Check electrolytes and glucose every hour after implementing IV insulin therapy.
- If the potassium is less than 3.2 mEq/dL, then replace it before starting insulin therapy while continuing hydration.
- Add dextrose to fluids once serum glucose is less than 250-300 mg/dL.
- Look for a source of infection with both hyper- and hypoglycemia.
- Consider early use of glucagon in the treatment hypoglycemia, especially when caused by oral agents.
- Pregnant women may have a glucose less than 200 mg/dL and still be in DKA.
- Make sure the nurse primes the IV tubing with the insulin solution. If not, a large amount of the insulin can bind to the plastic, thus decreasing the actual dose to a great degree.
- If a "crazy person" comes into the ED, check his or her glucose level.
- Hypoglycemia can mimic stroke symptoms, so don't forget the finger stick glucose test.
- Even a single dose of oral antidiabetic medication can be deadly to a pediatric patient.
- Captopril and sulfhydryl drugs will give a false positive result in a nitroprusside urine test.
- A venous blood gas is adequate to determine pH and HCO3.
References
1. Chiasson J-L, Aris-Jilwan N, Belanger R, et al. Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ 2003;168:859-866.
2. Fishbein H, Palumbo PJ. Acute metabolic complications in diabetes. In: National Diabetes Data Group, eds. Diabetes in America. 2nd ed. Washington, D.C.: National Institutes of Health; 1995:283-291.
3. Kitabchi AE, Fisher J, Murphy MB, et al. Diabetic ketoacidosis and the hyperglycemic hyperosmolar nonketotic state. In: Kahn CR, ed. Joslin's Diabetes Mellitus. Philadelphia: Lippincott Williams & Wilkins; 2005:738-770.
4. Malone M, Gennis V, Goodwin J. Characteristics of diabetic ketoacidosis in older verses younger adults. J Am Geriatr Soc 1992;40:1100-1104.
5. Matz R. Hyperosmolar nonacidotic diabetes (HNAD). In: Porte D JR, Sherwin RS, eds. Diabetes Mellitus: Theory and Practice. 5th ed. Amsterdam: Elsevier; 1997:845–860.
6. Leese GP. Frequency of severe hypoglycemia requiring emergency treatment in type 1 and type 2 diabetes: A population based study of health service recourse use. Diabetes Care 2003;.
7. Sugarman J. Hypoglycemia associated hospitalizations in a population with a high prevalence of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 1991;14:139-147.
8. Hattersley A. Unlocking the secrets of the pancreatic beta cell: Man and mouse provide the key. J Clin Invest 2004;114:314.
9. Pessin J, Saltiel A. Signaling pathways in insulin action: Molecular targets of insulin resistance. J Clin Invest 2000;106:165.
10. Saltiel A. Putting the brakes on insulin signaling. N Engl J Med 2003;349:2560.
11. Shi Y, Taylor S, Tan S, et al. When translation meets metabolism: Multiple links to diabetes. Endocr Rev 2003;24:91.
12. Roden M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol Sci 2004;19:92.
13. List J, Habener J. Glucagon-like peptide 1 agonists and the development and growth of beta cells. Am J Physiol Endocrinol Metab 2004;286(E):875.
14. [No Authors Listed.] Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1997;20:1183.
15. Genuth S, Alberti K, Bennett P, et al. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 2003;26:3160.
16. libman I, Pietropaolo M, Trucco M. Islet cell autoimmunity in white and black children and adolescents with IDDM. Diabetes Care 1998;21:1824-1827.
17. Alberti K, Zimmet P, consultation ftW. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabet Med 1998;15:539.
18. Umpierrez G, Casals M, Gebhart S, et al. Diabetic ketoacidosis in obese African-Americans. Diabetes Care 1996;44:790.
19. Pinhas-Hamiel O, Dolan L, Zeitler P. Diabetic ketoacidosis among obese African-American adolescents with NIDDM. Diabetes Care 1997;20:484.
20. Dussoix P, Vaxillaire M, Iynejian P, et al. Diagnostic heterogeneity of diabetes in lean young adults. Classification based on immunological and genetic parameters. Diabetes Care 1997;46:622.
21. Kitabchi AE, Umpierrez G, Murphy MB, et al. Managment of hyperglycemic crises in patients with diabetes. Diabetes Care 2001;24:131-153.
22. Siler SQ, Neese RA, Christiansen MP, et al. The inhibition of gluconeogenesis following alcohol in humans. Am J Physiol 1998;275:E897-E907.
23. Chiasson J-L, Josse R, Hunt J, et al. The efficacy of acarbose in the treatment of patients with non-insulin dependent diabetes mellitus. A multicenter controlled clinical trial. Ann Intern Med 1994;121:928.
24. Iwamoto Y, Kosaka K, Kuzuya T, et al. Effects of troglitazone: A new hypoglycemic agent in patients with NIDDM poorly controlled by diet therapy. Diabetes Care 1996;19:151.
25. Nolan J, Judvik B, Beerdsen P, et al. Improvment of glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med 1994;331:1188.
26. Yki-Jarvinen H. Drug Therapy: Thiazolidinediones. N Engl J Med 2004;351:1106.
27. Forman L, Simmons D, Diamond R. Hepatic failure in a patient taking rosiglitazone. Ann Intern Med 2000;132:118.
28. Al-Salman J, Arjomand H, Kemp D, et al. Hepatocellular injury in a patient recieving rosiglitazone. A case report. Ann Intern Med 2000;132:121.
29. Fonseca V, Valiquett T, Huang S, et al. Troglitazone monotherapy improves glycemic control in patients with type 2 diabetes mellitus: A randomized, controlled study. J Clin Endocrinol Metab 1998;83:169.
30. Fonseca V, Rosenstock J, Patwardhan R, et al. Effect of metformin and rosiglitazone combination therapy in patients with type 2 diabetes mellitus: A randomized controlled trial. JAMA 2000;283:1695.
31. Guan Y, Hao C, Cha D, et al. Thiazolidinediones expand body fluid volume through PPAR-gamma stimulation of ENa-Cmediated renal salt absorption. Nat Med 2005;11:861.
32. Bailey C. Biguanides and NIDDM. Diabetes Care 1992;15:755.
33. Bailey C, Turner R. Metformin. N Engl J Med 1996;334:574.
34. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;2001:1167.
35. Sirtori C, Pasik C. Re-evaluation of a biguanide, metformin: Mechanism of action and tolerability. Pharmacol Res 1994;30:187.
36. Bressler R, Johnson D. Pharmacological regulation of blood glucose levels in non-insulin-dependent diabetes mellitus. Arch Intern Med 1997;157:836.
37. Aguilar-Bryan L, Nichols C, Wechsler S, et al. Cloning the beta cell high-affinity sulfonulureas receptor: A regulator of insulin secretion. Science 1995;268:423.
38. Bressler R, Defronzo R. Drugs and diabetes. Diabetes Reviews 1994;2:53.
39. Weissberg-Benchell J, Antisdel-Lomaglio J, Seshadri R. Insulin pump therapy. Diabetes Care 2003;26:1079-1087.
40. Kelly A, Kyle E, McAlpine R. Venous pCO2 and pH can be used to screen for significant hypercarbia in emergency patients with acute respiratory disease. J Emerg Med 2002;22:15-19.
41. Kelly A, McAlpine R, Kyle. Agreement between bicarbonate measured on arterial and venous blood gases. Emerg Med Australas 2004;16:407-409.
42. Mcgarry J, Woeltje K, Kuwajima M, et al. Regulatin of ketogenesis and renaissance of carnitine palmitoyl transferase. Diabetes Metab Rev 1989;5:271-284.
43. Owen O, Trapp V, Skutches C, et al. Acetone metabolism during diabetic ketoscidosis. Diabetes 1982;31:242.
44. Slama G, Traynard P, Desplanque N, et al. The search for an optimized treatment of hypoglycemia. Carbohydrates in tablets, solution or gel for the correction of insulin reactions. Arch Intern Med 1990;150:589-593.
45. Brodows R, Williams C, Amatruda J. Treatment of insulin reactions in diabetics. JAMA 1984;252:3378-3381.
46. Bartlett D. Confusion, somnolence, seizures, tachycardia? Question drug induced hypoglycemia. J Emerg Nurs 2004;31:206-208.
47. McLaughlin S, Crandall C, McKinny P. Octreotide: An antidote for sulfonylurea-induced hypoglycemia. Ann Emerg Med 2000;36:133-138.
48. Scott C, Smith J, Cradock M, et al. Characteristics of youth-onset NIDDM and IDDM at diagnosis. Pediatrics 1997;100:84.
49. Dunger D, Sperling M, Acerini C, et al. ESPE/LWPES consensus statement on diabetic ketoacidosis in children and adolecents. Arch Dis Child 2004;89:188.
50. Adrogue H, Eknoyan G, Suki W. Diabetic ketoacidosis: Role of the kidney in the acid-base homeostasis re-evaluated. Kidney Int 1984;25:591.
51. Oh M, Carroll H, Uribarri J. Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis. Nephron 1990;54:1.
52. Edge J, Ford-Adams M, Dunger D. Causes of death in children with insulin dependent diabetes. Arch Dis Child 1999;81:318.
53. Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med 2001;344:264.
54. Rosenbloom A. Intracerebral crisis during treatment of diabetic ketoacidosis. Diabetes Care 1990;13:22.
55. Mahoney C, Vicek B, DelAguila M. Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr Neurol 1999;21:721.
56. Ramin K. Diabetic ketoacidosis in pregnancy. Obstet Gyenecol Clin North Am 1999;26:481.
57. Cullen M, Reece E, Homko C. The changing presentations of diabetic ketoacidosis during pregnancy. Am J Perinatol 1996;13:449.
58. Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000;13:4.
59. Montoliu J, Revert L. Lethal hyperkalemia associated with severe hyperglycemia in diabetic patients. Am J Kidney Dis 1985;5:47.
60. Besser G, Thorner M. Comprehensive Clinical Endocrinology, 3rd ed. Philadelphia: Mosby; 2002:324-335.
Diabetes is one of the most common chronic diseases in man, and as seen below, a frequent cause of emergency department (ED) visits. Diabetics now have more treatment options available, better delivery systems for their insulin and greater technology to allow them to self-monitor their glucose levels. We now know that good control of glucose is the best defense against the deadly long-term consequences of diabetesblindness, renal disease, peripheral neuropathy, etc.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.