Clinical Presentation, Evaluation and Management of Bleeding Disorders in Children
Clinical Presentation, Evaluation and Management of Bleeding Disorders in Children
Authors: Beng R. Fuh, MD, Assistant Professor of Pediatrics, Department of Pediatrics, Division of Pediatric Hematology and Oncology, Brody School of Medicine at East Carolina University; and Ronald M. Perkin, MD, Professor and Chairman, Department of Pediatrics, Brody School of Medicine at East Carolina University.
Peer Reviewer: Afshin Ameri, MD, Associate Professor of Pediatrics, Director of Pediatric Comprehensive Hemophilia Program, Medical College of Georgia, Augusta.
Bleeding is a common chief complaint in the emergency department.
Recognizing excessive bleeding and treating bleeding disorders may be challenging. Awareness of the differential, diagnostic strategies and treatment options is critical. This article presents a comprehensive approach to the patient with a bleeding disorder.
The Editor
Introduction
Appropriate hemostasis requires a complex interaction between platelets, vascular surface factors and clotting factors. The initial phase of hemostasis involves vascular reaction of the injured blood vessel and the formation of a platelet plug (primary hemostasis). Clotting factors then form the fibrin clot that provides the permanent seal (secondary hemostasis). A defect in any of the above factors can lead to abnormal bleeding. In addition, structural abnormalities such as hemangiomas or ectatic capillaries can lead to excessive bleeding. Bleeding disorders vary widely in severity and it can be challenging to establish which bleeding is excessive and which is within acceptable limits. It is not unusual for patients with congenital bleeding disorders to remain undiagnosed into their adulthood. Patients with excessive bleeding must be assessed for possible bleeding disorders and targeted treatment provided to prevent or minimize serious complications. Assessment should include a careful clinical history and laboratory evaluation. Disorders of primary hemostasis frequently present with epistaxis, bleeding of mucous membranes and bruising. Disorders of secondary hemostasis frequently present with soft tissue bleeds, hemarthrosis and large vessel bleeding. Management strategies vary from observation, local control, infusions of factor products to physical therapy and rehabilitation following bleeding complications.
Epidemiology and Etiology
Excessive bleeding secondary to primary defects in the coagulation system is rare and usually due to congenital quantitative or qualitative deficiencies in one of the components of the coagulation system. Patients can develop inhibitors following infections or medication leading to transient acquired bleeding disorders. The majority of cases of excessive bleeding in children are due to secondary causes such as vascular abnormalities or medication effects such as aspirin, ibuprofen, clopidrogel, etc. Inappropriate activation of the coagulation cascade can lead to platelet and coagulation factor consumption such as occurs in disseminated intravascular coagulation (DIC).
von Willebrand Disease (vWD) is the most common inherited bleeding disorder affecting about one to two percent of Americans.1,2 It is usually inherited in an autosomal dominant mode though some variants are autosomal recessive. von Willebrand antigen activates platelets by binding to the GP1b region of platelets. It also binds FVIII thereby stabilizing it. Very low levels of von Willebrand antigen can lead to low FVIII levels and inadequate formation of the platelet plug. vWD type IIb results in enhanced binding affinity of von Willebrand antigen to platelets resulting in increased platelet clearance and thrombocytopenia.3 Other variants of vWD are due to defects in the binding site to FVIII or absence of certain multimeric fractions. vWD can also arise from the development of autoimmune antibodies to the von Willebrand factor. Conditions associated with von Willebrand factor auto antibody formation include leukemia, SLE, and lymphoma.4 Wilms tumor5 and hypothyroidism6 have been associated with decreased synthesis of von Willebrand factor.
Hemophilia A (FVIII deficiency) has an incidence of 1 in 5,000 males. The gene for factor VIII is located on the X chromosome. Female carriers are usually unaffected, though extreme lyonization of the unaffected X chromosome, Turner's Syndrome, or daughters of affected fathers and carrier mothers can rarely lead to symptomatic females. Lyonization is the process whereby one X chromosome in each cell is randomly inactivated. It is described as severe when FVIII activity level is 1%, moderate if FVIII activity level is between 1% and 5%, and mild if FVIII is 5% to 40%.
Hemophilia B (FIX deficiency) is also X-linked and has an incidence of about 1in 40,000 males.
Both hemophilia A and B have no racial predilection, and about 30% of patients are due to new mutation with no prior family history of hemophilia.7
FXI deficiency, also referred to as hemophilia C, is inherited as an autosomal recessive condition. It is a very rare condition mostly found in people of Jewish ancestry.8
Other specific factor deficiencies include FXIII deficiency, FVII deficiency, hypofibrinoginemia, dysfibrinoginemias, etc.
Vitamin K deficiency leads to a decrease in vitamin-K-dependent coagulation factors and can lead to bleeding. Maternal use of anti-convulsants are a common cause of early-onset hemorrhagic disease of the newborn. Late-onset hemorrhagic disease of the newborn usually occurs in exclusively breastfed infants with inadequate neonatal vitamin K prophylaxis. In older children, vitamin K deficiency can result from malabsorption syndromes, prolonged emesis, and warfarin use.
Liver disease causes a decrease in the synthesis of clotting factors, the most sensitive of which are FV and FVII.
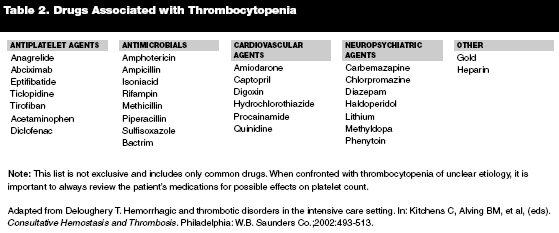
Thrombocytopenia secondary to congenital amegakaryocytic thrombocytopenia, thrombocytopenia absent radius (TAR), etc. are rare. Fanconi anemia and other forms of aplastic anemia are other rare causes of thrombocytopenia. Peripheral consumption or destruction or bone marrow infiltration such as occurs in leukemia can also lead to thrombocytopenia. Table 1 shows the common causes of thrombocytopenia, and Table 2 shows drugs frequently associated with thrombocytopenia.9
 |
 |
The Coagulation Cascade
Most coagulation factors are synthesized in the liver. Factors II, VII, IX, X, and XII are synthesized as inactive enzymes that become active when cleaved. Factors II, VII, IX, and X are vitamin K dependent. Activation of coagulation factors culminates in the generation of thrombin, which in turn cleaves fibrinogen to produce fibrin and activates platelets. Figure 1 shows a scheme of the coagulation cascade.
Clinical Presentation
Clinical presentation varies depending on the etiology of the bleeding disorder. It can be difficult to differentiate between normal bleeding and excessive bleeding. A very detailed bleeding history, including a family history, is critical in the diagnosis of a bleeding disorder. If there is a family history of excessive bleeding, a pedigree should be developed to identify a possible hereditary disease. Disorders of primary hemostasis usually present with epistaxis, mucosal membrane bleeding, and superficial ecchymosis, while disorders of secondary hemostasis usually present with joint bleeding, intramuscular bleeding, or other large-vessel bleeding.
Thrombocytopenia. Thrombocytopenia usually manifests itself as petechiae, ecchymoses, and mucous membrane bleeding. There can also be excessive immediate bleeding following surgery or trauma. Bleeding is rare at platelet counts greater than 50,000/mm3, in the absence of a platelet dysfunction. The risk of bleeding is also higher in disorders of decreased production, such as chemotherapy-induced thrombocytopenia, than in disorders of increased platelet destruction such as ITP. The PT and aPTT are usually normal.
von Willebrand Disease. Most patients with vWD are asymptomatic. When symptomatic, it is usually as mucocutaneous bleeding such as menorrhagia in females, epistaxis, or ecchymosis. Occasionally, patients may present with excessive post-surgical bleeding, such as post tonsillectomy. Type III vWD may present with symptoms similar to hemophilia A or B. Since most forms of vWD are inherited in an autosomal dominant fashion, a good family history often reveals other family members with symptoms of excessive bleeding. APTT is usually prolonged, but may be normal, especially in mild vWD. von Willebrand factor (vWF) is an acute phase reactant, and even the stress of impending phlebotomy may be enough to increase vWF levels.10
Hemophilia A. Patients can present with a wide range of bleeding manifestations, from easy bruising to joint bleeds to intracranial bleeding. Recurrent joint bleeding can lead to target joints with the risk of permanent injury. In the neonatal period, hemophilia A can present as excessive post-circumcision bleeding, excessive bleeding post-venipuncture, or intracerebral hemorrhage. Affected individuals are usually male, and there is usually history of affected males in the maternal family. Rarely, females may be affected secondary to extreme lyonization, Turner's Syndrome, daughter of a carrier mother, or daughter of a hemophiliac father and carrier mother.
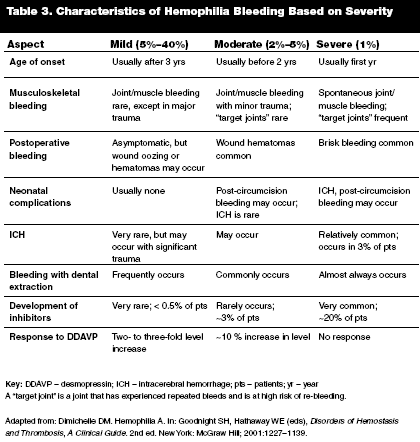
Hemophilia B. Hemophilia B has a similar presentation as hemophilia A. Table 3 summarizes the common complications of hemophilia based on severity.
 |
Factor XIII Deficiency. Factor XIII deficiency may present with bleeding soon after birth, or as delayed post-surgical bleeding; for example, 24–72 hours post-tonsillectomy.
Factor XI and XII. Factor XI and XII deficiencies are usually asymptomatic, or there may be mild bleeding.
Disorder of fibrinogen. Disorders of fibrinogen may manifest in different ways depending on severity, as shown in Table 4.
 |
Vitamin K deficiency. Vitamin K deficiency can present as early- or late-onset hemorrhagic disease of the newborn, with intracerebral hemorrhage, cephalohematoma, etc. Older children can present with mild to severe bleeding; PT and aPTT are usually prolonged.
Disseminated Intravascular Coagulation (DIC). Clinical symptoms of DIC may be masked by the underlying disease, making a high level of suspicion and evaluation necessary. Bleeding diathesis is usually more pronounced than micro-thrombosis. Oozing from intravenous catheter sites, ecchymosis, and petechiae are frequent manifestations.
Evaluation
When confronted with acute or chronic bleeding, it is important to determine if the bleeding is within normal limits or is excessive. Evaluation should include a good history, including details on the locations, amount and frequency of bleeding, physical examination, and basic laboratory tests. Based on these findings, further testing may be done. Questions should be specific, and patients and parents should describe bleeding qualitatively and quantitatively, avoiding the terms "normal" or "abnormal." Parents often base "normal" or "abnormal" on their own experiences; for example, a mother with menorrhagia secondary to undiagnosed vWD may describe excessive menstrual flow in her daughter as normal. Table 5 is a suggested hemostatic history questionnaire.11 Pictorial charts can be used to assist in assessing the amount of menstrual bleed.12
 |
Physical examination can give very important clues as to the underlying disorder. Petechiae usually indicate thrombocytopenia. Enlarged spleen and or enlarged liver may signify chronic illness or malignancy. Hyperextensibility of joints may indicate a connective tissue dysfunction, as in Ehlers-Danlos syndrome. Hemangiomas may suggest a consumptive etiology; joint abnormalities may suggest hemophilia.
Screening tests. If a bleeding abnormality is suspected, the following tests should be obtained. Attention must be paid to proper collection of samples including the right plasma to reagent volume and the avoidance of heparin contamination. Reference values are age dependent and vary from laboratory to laboratory which is a function of reagents used. It is important to have this in mind when interpreting results. For example aPTT is usually prolonged in neonates. Most bleeding factors reach adult levels by age 6–12 months.
Complete blood count (CBC). This will determine if there is a quantitative platelet disorder and if only platelets are affected. In bone marrow failure syndromes or infiltrative marrow processes, there are often abnormalities in other hematopoietic cell lines. The size of platelets should also be assessed. Large platelets often denote an active bone marrow as seen in processes associated with peripheral platelet destruction such as occurs in ITP. Normal platelets are seen in marrow suppression or failure and small platelets may be seen in Wiskott Aldrich Syndrome (WAS).
Prothrombin Time (PT)/International Normalized Ratio (INR). This evaluates the extrinsic pathway of the coagulation cascade. Factor VII is unique to the extrinsic pathway so a factor VII deficiency would result in a prolonged PT and normal aPTT. INR normalizes the variability in the sensitivities of thromboplastin reagents to low concentrations of some coagulation proteins. It is normally used to monitor oral anticoagulant therapy.
Activated Partial Thromboplastin Time (aPTT). Assesses the intrinsic pathway of the coagulation cascade. The aPTT is usually normal until factors levels drop to less than 30%.
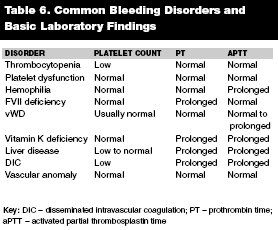
Table 6 shows the common differential diagnoses based on platelet count, PT, and aPTT findings.
 |
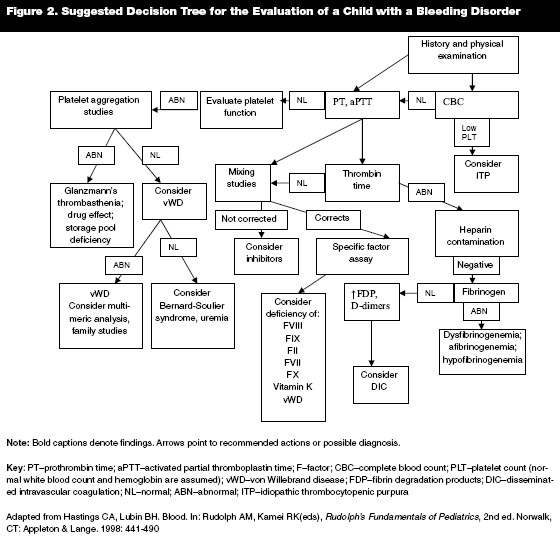
If the above findings suggest a disorder of hemostasis, specific testing should be done. Figure 2 shows a suggested decision-making tree for the evaluation of a hemostatic disorder.13
 |
Idiopathic thrombocytopenic purpura (ITP). If a CBC shows isolated thrombocytopenia, ITP is very likely, especially if the smear reveals large platelets. The patient should be managed presumptively as ITP, and no further testing is necessary.
von Willebrand disease. von Willebrand antigen and activity levels can fluctuate and repeat testing is frequently necessary. Individuals with blood group O generally have lower vWF levels. A discrepancy between von Willebrand antigen level and activity level (ristocetin cofactor) may suggest a qualitative vWD subtype. In this case, multimeric analysis and platelet aggregation studies are important. Care should also be taken when interpreting von Willebrand panel results in patients who are on birth control, as this may artificially increase the level of von Willebrand antigen level. von Willebrand antigen level is a quantitation of vWF using immunoelectrophoresis or ELISA.14 Ristocetin induces the binding of vWF to the glycoprotein Ib receptor of platelets; this is used to measure the activity of vWF. vWF carries FVIII in circulation, thereby protecting it from degradation, and its level is helpful in diagnosing vWD. vWF is composed of different multimeric sizes, and these can be separated via gel electrophoresis. The distribution is normal in type I vWD, while type IIA vWD has a decrease in large multimers.
Management of Bleeding Disorders
In cases of active bleeding priority has to be controlling the bleeding and maintaining the airway and circulation. Where possible, compression and pressure should be applied, with caution taken to limit damage to vital structures. If the patient has a known specific hemostatic defect, specific treatment should be administered as soon as possible. Generally patients with established hemophilia should carry an alert bracelet bearing their diagnosis. If the patient is hemodynamically unstable, consider transfusion with packed red blood cells (PRBC), platelets, and fresh frozen plasma (FFP).
Hemophilia. Patients with known severe hemophilia need to be treated promptly whenever CNS bleeding is suspected or possible for example, after head trauma even if there is no obvious sign of bleeding. Treatment should be administered prior to imaging studies.
Patients who develop bleeding secondary to other conditions need treatment for the primary condition in addition to managing active bleeding. Avoid intramuscular (IM) injections. In patients with mild hemophilia A, Desmopressin (DDAVP) 0.3 mcg/kg may be used if the patient is known to be a responder. Desmopressin causes the release of vWF and FVIII from storage sites in endothelial cells, thereby raising serum FVIII levels. DDAVP can raise FVIII levels by up to six-fold.14 However, it is difficult to predict serum levels of FVIII after the administration of DDAVP, especially in stress situations. DDAVP should therefore be used only before the patients arrives at the emergency department or when IV access cannot be established for factor concentrate administration. If repeat dosing is necessary, monitor factor VIII levels for response and monitor for tachyphylaxis and sodium levels. Avoid treating more often than every eight hours or using more than three doses per treatment course.14
Patients with moderate or severe hemophilia should be treated with FVIII or FIX products for hemophilia A and hemophilia B, respectively. Table 7 shows suggested dosing guidelines for patients with factors VIII and IX deficiency for different types of bleeds. Wherever possible, recombinant products should be used because these have lower treatment risks.
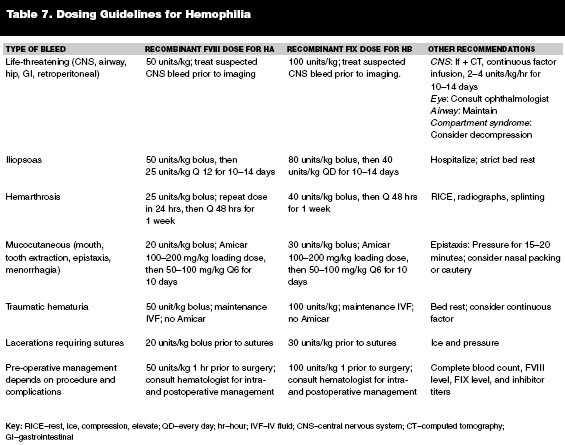 |
Hemophilia patients with inhibitors. For patients with hemophilia A with inhibitors and non-life- or limb-threatening bleeding, high-dose FVIII (100 units/kg bolus followed by 20 units/kg/hr via continuous infusion should be administered). If bleeding persists, treat with activated FVII (Nvo 7, Novo Nordisk). If bleeding is not controlled by FVII, consider prothrombin complex concentrates (PCC) at 75 units/kg.15 Monitor closely for DIC and thrombosis, and a hematologist should be consulted. Porcine FVIII is available for use in patients with inhibitors, but should only be used under the guidance of the hematologist.
For patients with hemophilia B with inhibitors, activated FVII at a dose of 100 mcg/kg (range 90–240mcg/kg) every 6 hours (range 2–12 hours) should be used. If bleeding persists, consider PCC at a dose of 75 units/kg and monitor closely for DIC and thrombosis.15 PCC should not be given to patients with a history of anaphylaxis to FIX. The hematologist should be consulted.
Important facts about factor FVIII and FIX replacement products. FVIII is available as recombinant products (recombinate, kogenate, helixate, advate, refacto, etc.), monoclonal purified products (hemofil-M, Monoclate, AHF-M), and porcine factor (Hyate-C). Whenever possible, recombinant factors should be used; it has a half life of 8–12 hours, and a 1 unit/kg dose increases plasma FVIII level by 2%. It is the treatment of choice in hemophilia A.
Recombinant FIX is available as recombinant products (benefix) or highly purified products (monomine, alphanine, etc.). It has a half life of 18–24 hours and a 1 unit/kg dose increases plasma FIX level by 0.75%–1 %. It is used to treat hemophilia B.
Factor VII is available as a recombinant product. It has a half life of two hours and is used to treat FVII deficiency or hemophilia A or B patients with inhibitors.
Amicar (epsilon-aminocaproic acid) is an anti-fibrinolytic agent that helps stabilize the fibrin clot. Dosing is 100–200 mg/kg (max 10 g) loading dose followed by 50–100 mg/kg (max 5 g) every 6 hours, not to exceed 30 g/day. It is most useful for mucous membrane bleeding, especially nasal, oral, and gastrointestinal tract bleeding, where enzymes contained in secretions can accelerate clot degradation. It can be administered orally or parenterally and should be given every six hours for 7–10 days. For oral bleeding, it can be used as swish and spit. Swish for two minutes prior to expectoration. Patient should not eat or drink anything (remain NPO) at least one hour after swish and spit. Amicar is contraindicated in patients with hematuria.
Most patients with hemophilia receive comprehensive care at specialized regional centers, and studies have shown that this leads to significantly improved outcomes. Hemophilia treatment centers generally have around-the-clock coverage to assist patients and parents, emergency department, and primary care physicians in case of emergency. Patients may carry a supply of their factor, and this can be used in case the treating physician does not have readily available factor products.
Von Willebrand Disease. Desmopressin (DDAVP). DDAVP acts by releasing vWF from storage sites in endothelial cells. This leads to a rise in serum levels of vWF and FVIII. Use 0.3 mcg/kg diluted in 25–50 mL normal saline infused over 30 minutes or nasal spray. DDAVP should be used with caution in patients with vWD type 2B, since it can worsen thrombocytopenia in this subset of patients. (See Table 8.)
 |
Nasal DDAVP for vWD (Stimate) should not be confused with generic DDAVP used for enuresis or panhypopituitarism.
DDAVP dosing. For body weight < 50kg, give 1 puff DDAVP (150 mcg). For weight > 50kg, give 2 puffs (300 mcg). vWF has a half life of 8–12 hours.
Stimate is appropriate for home use and minor bleeding only. In the emergency department setting, cryoprecipitate or vWF-enriched plasma preparations should be used. Severe bleeding can also be treated with Humate-P or alphanate at 25 ristocetin cofactor units/kg.
Other treatment products. FFP. Use for disorders for which factor concentrates are not available. Dosing is 5–10 mL/kg; 1 mL of FPP ~ 1 unit of each clotting factor. For doses > 250 mL, consider one single donor plasmapheresis product to limit donor exposure and risk of infection.
Cryoprecipitate. Use for afibrinogenemia, hypofibrinogenemia, dysfibrinogenemia, factor XIII deficiency in situations where FFP is contraindicated due to volume overload concerns. One donor unit contains ~100 units of factor VIII and vWF and ~ 1500mg of fibrinogen. Cryoprecipitate has a higher risk of HIV and/or hepatitis C transmission relative to FFP and virally inactivated or recombinant FVIII and vWF concentrates.
Fibrogammin P (FXIII concentrate). May be considered for FXIII deficiency.
Fibrin glue. May be used for small to moderate lacerations and dental extraction but this should be done with caution, as it may lead to the development of thrombin inhibitors.
Special Considerations. Epistaxis, Epistaxis is common in children and may or may not be related to a disorder of hemostasis. Management should involve the following:
Place patient in sitting position to decrease venous pressure. If patient cannot sit, place head higher than heart and turn head to the side.
If possible, flex the patient's neck forward with the chin touching the chest.
Firmly compress the lower part of the nose for 20 minutes.
If bleeding continues, reassess location of compression. If bleeding recurs, reapply pressure for 20 minutes.
If the patient has a known hemostatic disorder, administer specific treatment as outlined previously.
Patient should avoid blowing the nose for at least 12 hours to avoid dislodging the clot.
Consider nasal packing if bleeding is persistent and profuse. If packing is applied, consider humidification or nasal saline sprays to prevent drying of the nasal mucous membranes. Also consider broad-spectrum antibiotics with good coverage for skin flora in immune-compromised patients. Packing may be left in place for up to five days.
If the area of bleeding can be identified and is circumscribed, consider cauterization.
If significant blood loss occurs, consider transfusion of PRBC.
Obtain a basic hemostasis laboratory evaluation and consider evaluation by an ear, nose and throat specialist.
Menorrhagia. The most frequent cause of menorrhagia in children is dysfunctional uterine bleeding, but these can frequently be exacerbated by an undiagnosed underlying mild or moderate vWD. Some studies have shown that up to 20% of women with menorrhagia may have vWD.16 Immediate treatment should be aimed at controlling bleeding and stabilizing the patient. High-dose estrogen until bleeding ceases followed by a taper then regular oral contraception is usually helpful. If the patient has a diagnosed bleeding disorder, specific treatment should be administered. Patients with significant bleeding may need PRBC transfusion. An evaluation for vWD should be initiated in patients with menorrhagia. Oral contraceptives can increase serum vWF levels and in mild vWD, this is often adequate to allow for allow coagulation.
ITP. Most cases of ITP resolve spontaneously and never need intervention. If the platelet count is less than 10,000/mm3, treatment with IVIG 0.8–1 g/kg should be considered. The dose may be repeated after 24 hours. A response should be expected in 24–48 hours. Other treatment options include steroids, plasmapheresis, rituximab, and other chemotherapeutics. Before administering steroids, careful evaluation should be done to exclude leukemia as a cause of thrombocytopenia. Avoid platelet transfusions, as these are likely to be destroyed by the underlying immune process.
Patients with ITP rarely bleed, but if central nervous system bleeding or other major bleeding occurs, an emergent splenectomy and continuous platelet infusion should be considered.
CNS complications. These can be devastating and range from subtle cognitive defects to debilitating strokes. This underscores why any suspected CNS bleed must be treated promptly.
Hemarthrosis. In addition to acute hemarthrosis, which is characterized by significant swelling, warmth, erythema, pain, and a decrease in range of motion that improves with treatment with specific factor, chronic hemarthrosis occurs in target joints, resulting in persistent limitation in the range of motion with little swelling or erythema. Chronic hemarthrosis may require splinting, prophylactic factor treatment, and physical therapy.
Some long-term complications are treatment-related and range from the development of inhibitors to line infections and HIV infection. Table 9 shows relative risks of certain complications of various factor replacement products. The risk of transmitting viral infections such as hepatitis, slow viruses, and HIV has continuously decreased as highly purified and recombinant factor replacement products have become the mainstay of treatment.
 |
Summary
A bleeding child can present a diagnostic and management challenge. When confronted with an acutely bleeding child, immediate management should involve airway management, breathing, and circulation. A detailed assessment to determine the source and extent of bleeding as well as patient and family history should guide further management. Because the possible etiologies of bleeding in children are diverse, resulting in a period of several weeks to arrive at a specific diagnosis, management should be based on probable etiologies. Whenever a life threatening bleed such as CNS bleeding is suspected, treatment should be prompt and not delayed for diagnostic studies. Regional hemophilia treatment centers have specialists available around the clock to assist in the management of hemophilia patients. Having a high index of suspicion for disorders of hemostasis not only helps in establishing specific diagnosis and in appropriate management in case of trauma or surgery, but can avoid unnecessary procedures such as hysterectomies for excessive menorrhagia. Anyone diagnosed with hemophilia should be referred to a comprehensive hemophilia treatment center. With appropriate management, children with bleeding disorders can lead normal lives with few complications.
References
1. Kashyap AS, Anand KP, Kashyap S, et al. Treatment of von Willebrand Disease. N Engl J Med 2004;351:2345–2346.
2. Montgomery RR. Von Willebrand Disease. In: Goodnight SH, Hathaway WE (eds), Disorders of Hemostasis and Thrombosis, A Clinical Guide. 2nd ed. New York: McGraw Hill 2001:115–125.
3. Randi AM, Rabinowitz I, Mancuso DJ, et al. Molecular basis of von Willebrand disease type IIB, Candidate mutations cluster in one disulfide loop between proposed platelet glycoprotein 1b binding sequences. J Clin Invest 1991;87:1220–1226.
4. Montgomery RR, Gill JC, Scott JP. Hemophilia and von Willebrand disease. In : Nathan DG, Orkin SH (eds), Nathan and Oski's Hematology of Infancy and Childhood. 5th ed. Philadelphia: WB Sauders, 1998: 1631–1659.
5. Scott JP, Montgomery RR, Tubergen DG, et al. Acquired von Willebrand's disease in association with Wilm's tumor: Regression following treatment. Blood 1981;58:665–669.
6. Galli-Tsinopoulou A, Stylianou C, Papaionnou G, et al. Acquired von Willebrand's Syndrome resulting from untreated hypothyroidism in two prepubertal girls. Haemophilia 2006;12:675–676.
7. Dimichelle DM. Hemophilia A. In: Goodnight SH, Hathaway WE (eds), Disorders of Hemostasis and Thrombosis, A Clinical Guide. 2nd ed. New York: McGraw Hill 2001:1227–139.
8. Kadir RA, Kingman CE, Chi C et al. Screening for factor XI deficiency amongst pregnant women of Ashkenazi Jewish Origin. Haemophilia 2006;12:625–628.
9. Deloughery T. Hemorrhagic and thrombotic disorders in the intensive care setting: In: Kitchens C, Alving BM, Kessler C (eds). Consultative Hemostasis and thrombosis. Philadelphia: WB Saunders Company 2002:493–513.
10. Casonato A, Pontara E, Bertomoro A, et al. Fainting induces an acute increase in the concentration of plasma factor VIII and von Willebrand factor. Haematologica 2003;6:688–693.
11. Goodnight SH, Hathaway WE (eds), Evaluation of bleeding tendency in the outpatient child and adult In: Disorders of Hemostasis and Thrombosis, A Clinical Guide. 2nd ed. New York: McGraw Hill 2001:52–60.
12. Higham JM, O'brien PMS, Shaw RW. Assessment of menstrual blood loss using a pictorial chart. Br J Obstet Gynaecol 1990;97: 734–739.
13. Lubin BH. Blood. In: Rudolph AM, Kamei RK(eds), Rudolph's Fundamentals of Pediatrics, 2nd ed. Norwalk, CT: Appleton & Lange 1998:441–490.
14. Fogarty PF, Rick ME. Disorders of hemostasis II. In: Rodgers GP, Young NS (eds), Bethesda Handbook of Clinical Hematology. Philadelphia: Lippincott Williams & Wilkins 2005:280–293.
15. Hastings C. The Children's Hospital of Oakland Hematology/Oncology Handbook. St Louis, Mosby, 2002:88
16. Kujovich JL. von Willebrand disease and menorrhagia: Prevalence, diagnosis and management. Am J Hematol 2005;79(3): 220–228.
Bleeding is a common chief complaint in the emergency department. Recognizing excessive bleeding and treating bleeding disorders may be challenging. Awareness of the differential, diagnostic strategies and treatment options is critical. This article presents a comprehensive approach to the patient with a bleeding disorder.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.