Pediatric Hematologic Emergencies
Teng Lu, MD, Emergency Physician, Kaiser Permanente, West Los Angeles
N. Ewen Wang, MD, Professor of Emergency Medicine, Associate Director Pediatric Emergency Medicine, Stanford School of Medicine, Stanford, CA
Peer Reviewer
Ronald M. Perkin, MD, MA, Professor and Chairman, Department of Pediatrics, The Brody School of Medicine at East Carolina University, Greenville, NC
Statement of Financial Disclosure
Dr. Lu (author), Dr. Wang (author), and Dr. Perkin (peer reviewer) report no financial relationships relevant to this field of study.
Hematologic abnormalities are commonly encountered in the pediatric emergency department (ED). Although children and adults can have similar hematologic abnormalities (e.g., iron deficiency anemia), other disease entities are predominantly pediatric (e.g., hemolytic uremic syndrome) or present differently in children than in adults (e.g., immune thrombocytopenia). In addition, children can present to the ED with acute exacerbations of previously undiagnosed congenital diseases such as sickle cell anemia.
This article will cover disorders of the hemogram: red blood cells, platelets, and white blood cells. The first part will contain a review of the normal production of cell lines. The second part will include discussions of abnormalities in each cell line, starting with a brief introduction and discussion of relevant history and physical exam, followed by highlights on notable conditions. Bleeding disorders, such as hemophilia and Von Willebrand disease, and specific types of leukemias/lymphomas are out of the scope of this article and will not be discussed extensively. Overall, the ED physician must be comfortable with a spectrum of diseases, ranging from common conditions, such as iron-deficiency anemia, to largely benign conditions, such as immune thrombocytopenic purpura, to life-threatening conditions, such as leukostasis or hemolytic anemia.
— Ann M. Dietrich, MD, FAAP, FACEP, Editor
Introduction
Circulating blood cells are produced in the bone marrow (see Figure 1). Many hematologic disorders may be characterized by either elevated or diminished levels of one or multiple cell lines. These can be categorized by mechanism as disorders due to decreased production (e.g., marrow failure), disorders due to increased destruction (e.g., hemolysis), or addition to poorly functioning cells (e.g., leukocytes in malignancy). Understanding the underlying mechanism of disease can be critical to management (see Table 1).
Figure 1. Overview of Bone Marrow Hematopoiesis |
|
Platelets are broken off from megakaryocyte precursors. Reticulocytes are immature erythrocytes. Bands (not shown) are immature neutrophils.
|
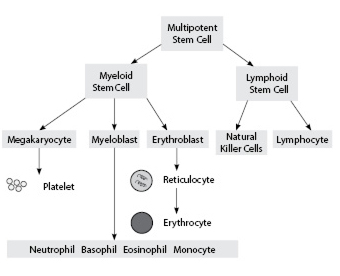
Table 1. Differential Diagnosis of Red Blood Cell, Platelet, and Granulocyte Disorders Categorized by General Mechanism |
|||
|
Normal lifespan |
110-120 days |
8-10 days |
4-30 days |
|
Disordered production |
|
|
|
|
Increased destruction |
|
|
|
|
|
Red Blood Cells |
Platelets |
Leukocytes |
|
Normal values (variable by age) |
11-14 g/dL |
150,000-450,000/uL |
4,000-11,000/uL |
Anemia and Polycythemia
Interpreting the Hemogram
General understanding of how to interpret the hemogram is a critical skill for the ED physician. Normal values for pediatric hemoglobin level vary by age (see Table 2). In the newborn, high hemoglobin concentrations in the 16.5-18.5 g/dL range is normal. In the first 2 months of life, “physiologic anemia” with hemoglobin levels as low as 10-11 g/dL can be seen due to a decrease in erythropoietin production in response to the rise in arterial oxygen level after birth. After this period, a steady rise in hemoglobin should occur, reaching a peak of 14.0 g/dL around age 14 years.2,3
Table 2. Pediatric Hemoglobin and MCV Level by Age2,3 |
|
|
Age |
Hemoglobin level (g/dL) |
|
Birth |
16.5-18.5 |
|
2-6 months |
11.2-12.6 |
|
6 months to 12 years |
12.0-13.5 |
|
12-18 years |
14.0-14.5 |
|
Age |
MCV (fL) |
|
Birth |
98-116 |
|
1 month to 9 years |
(70 + age) - 96 |
|
9-18 years |
80-96 |
The mean corpuscular volume (MCV), measured in femtoliters (fL), is the average volume of red blood cells. Like pediatric hemoglobin levels, the MCV also varies by age. The MCV is perhaps the most useful parameter for the ED physician in the evaluation of anemia, as it can help narrow the differential diagnosis (see Table 3).
The blood smear can provide additional clues to the etiology. For example, central pallor (hypochromic) in microcytic red cells is commonly seen in iron deficiency and thalassemias. Spherocytes can be seen in spherocytosis or hemolysis. Fragmented or bite cells, schistocytes, and Heinz bodies are all indicative of hemolytic processes. Abnormalities in other cell lines, such as white blood cell blasts, should raise suspicion for leukemia or lymphoma.
The reticulocyte count is another useful parameter. Reticulocytes are young, immature red cells that contain residual ribonucleic acid. At birth, the reticulocyte count can be as high as 3-7%, falling to 0.1-1% shortly after birth due to erythropoietin suppression. After this time and through adulthood, a normal reticulocyte count ranges between 1-2%. Reticulocytosis > 3% indicates active erythropoiesis, which can be seen in hemolysis or blood loss. In contrast, anemia with normal or low reticulocytes indicates a poor bone marrow response, which can be seen in aplastic anemia, marrow aplasia, or toxin exposure (see Table 3).
Table 3. Differential Diagnosis of Anemia Categorized by MCV |
||
|
Microcytic (MCV < 70 + age*) |
Normocytic (MCV = 70 + age* to 96) |
Macrocytic (MCV > 96) |
|
Problems with Hbg synthesis Iron deficiency Thalassemia Sideroblastic Drugs/toxins (lead, isoniazid) |
Hemolytic (retic > 3%) Autoimmune hemolytic anemia Hemoglobinopathies Enzymopathies (G6PD deficiency pyruvate kinase deficiency) Membranopathies (spherocytosis, elliptocytosis) Hemolytic uremic syndrome Disseminated intravascular coagulation Artificial heart valve Non-hemolytic (retic < 3%) Anemia of chronic disease Acute blood loss Chronic renal failure Hypersplenism |
Megaloblastic B12 or folate deficiency Purine or pyrimidine deficiency Drugs (methotrexate, anticonvulsants) Non-megaloblastic Aplastic anemia Myeloblastic syndrome Hypothyroidism Liver disease Down syndrome |
|
(*age up to 9 years old) |
||
History and Physical Exam
In the hemodynamically unstable patient with lethargy, mottling, hypoxia, hypotension, or other signs of hypoperfusion, initial ED management should include obtaining intravenous access and stabilizing airway, breathing, and circulation. Blood loss anemia should be ruled out first, then ED physicians should consider other differential diagnoses, such as disorders of erythrocyte synthesis or hemolysis. Type and crossed blood products are preferred, but in emergent situations O negative blood can be given. In general, packed red blood cells are transfused at aliquots of 10 mL/kg, each of which should raise hemoglobin level by 2 g/dL. The rate of transfusion can be over minutes to 4 hours based on the clinical situation.
In stable patients, evaluation of pediatric anemia begins with a thorough history and physical exam. Key components of history include family history of blood dyscrasias, episodes of neonatal jaundice, occurrence of prior similar events, exposure to new medications, or potential sources of blood loss. Other important questions include travel history, home conditions, dietary habits, and constitutional symptoms (e.g., fever, night sweats, or weight loss). Notable physical exam findings include nail bed or conjunctival pallor, scleral icterus or jaundice, oral ulcers (indicative of inflammatory bowel disease), glossitis or stomatitis, murmurs, as well as abdominal masses and hepatosplenomegaly.
Iron Deficiency Anemia
Iron deficiency anemia is the most common cause of anemia in early childhood. In the United States, approximately 9% of children ages 1-3 years have iron deficiency, and 2-3% have iron deficiency anemia.4 In low-income populations, prevalence can be as high as 14%.5 The hemogram in iron deficiency anemia classically shows a low hemoglobin level, low MCV, and low reticulocyte count. The red cell distribution width can be elevated.
Common causes of iron deficiency anemia in children include insufficient intake, decreased absorption, consumption of non-formula cow’s milk before 12 months of age, and occult gastrointestinal blood loss due to cow’s milk protein colitis. Daily requirements of iron are 1 mg/kg (maximum 15 mg) for term infants and 7-10 mg daily for toddlers. Iron-enriched formulas contain high iron concentrations (12 mg/L) but have low bioavailability (4-6%). On the other hand, breast milk contains lower iron concentrations (1 mg/L) with high bioavailability of 50%.6 Other sources of iron include iron-fortified cereals, vegetables, fish, and meat. Vitamin C is important in enhancing iron absorption in the diet.
Prevention and parental counseling is critical in the management of pediatric iron-deficiency anemia. In exclusively breastfed term infants, iron supplementation should be started after 4 months. Formula-fed infants generally do not need additional supplementation due to adequate intake for fortified formulas. Other sources of iron, such as iron-fortified cereals, pureed meats, and vitamin C sources, should be introduced in the diet after 6 months. Toddlers between ages 1-3 years should have at least three servings of iron-rich foods daily. Milk should be limited in this group to a maximum of 600 mL (20 ounces) daily, as excess consumption can lead to decreased intake of other foods (see Table 4).
Table 4. General Recommendations for the Prevention and Treatment of Iron Deficiency Anemia in Children7 |
|
|
Preterm infants |
Breastfed: begin supplementation after 2 weeks of age Iron-fortified formula fed: no additional supplementation |
|
Term infants |
Breastfed: begin supplementation at 4 months of age Iron-fortified formula fed: no additional supplementation |
|
4-6 months |
Introduce iron-containing foods (iron-fortified cereals, pureed meats, vitamin C sources) |
|
1-3 years old |
No whole cow’s milk until 1 year of age After 1 year of age, milk consumption to 600 mL (20 ounces) daily Limit to 3 servings of iron-containing foods daily |
|
Iron supplementation dosing (5 mg ferrous sulfate = 1 mg elemental iron) |
|
|
Ferrous sulfate |
3 mg/kg once to twice daily, orally, between meals with juice |
Treatment for iron-deficiency anemia in children is in the form of ferrous sulfate, in addition to dietary counseling and modification. A repeat hemoglobin level will need to be checked after 1 month to assess for response. Transfusion is only indicated in patients with tachypnea, tachycardia, lethargy, or other manifestations of symptomatic anemia.7 In well-appearing children, transfusion is rarely necessary, even with low hemoglobin levels of 4-5 g/dL, as long as follow-up can be ensured. However, some physicians consider hemoglobin levels of 5 g/dL as a threshold for admission for observation or transfusion. Children requiring transfusions should be admitted to the hospital and transfused at slow rates of 5-10 cc/kg over 4 hours.
Lead Poisoning
The “toxic” lead level is determined by the blood lead level (BLL) seen in 97.5% of U.S. children. This is currently > 5 mcg/dL. Overall, the incidence and severity of lead poisoning in children is decreasing due to increased public awareness, with 77.8% of children in the United States having levels > 10 mcg/dL in the 1970s compared to 1.6% in 2002.8 Despite this progress, children from lower socioeconomic groups, recent immigrants, or those living in poor inner-city areas still have a disproportionately higher risk of lead poisoning.
Many children with lead poisoning are asymptomatic. When symptoms do arise, they often are neurologic, hematologic, or gastrointestinal. Irritability, hyperactivity, peripheral neuropathy (e.g., hearing loss), and insomnia can be seen at BLL > 10 mcg/dL. Encephalopathy (e.g., persistent vomiting, seizures, altered mental status) can occur at BLL > 100 mcg/dL. Hematologically, lead interferes with hemoglobin synthesis, which can result in microcytic anemia at BLL > 40 mcg/dL. Common gastrointestinal symptoms include abdominal pain, vomiting, and constipation.
Suspicion for lead toxicity should warrant collecting a detailed environmental history. Homes built before the 1980s pose a risk due to lead-based paint or plumbing. Housing proximity to industrial sites is also a red flag. Other risk factors include access to lead-glazed ceramics, stained glass, and imported crayons, toys, or cosmetics. Diagnosis is made by obtaining venous blood lead levels; results are not often available in the ED. If anemia is detected, it is also important to send for a full iron studies panel, including total iron-binding capacity, transferrin saturation, ferritin, and reticulocyte count. If there is a history of pica or if the child has acute encephalopathy of unclear origin, abdominal radiograph can be useful to screen for lead flecks. In chronic exposure, long bone radiographs may show lead lines (bright opaque lines lining the edge of long bone metaphyses).
At blood lead levels between 5-45 mcg/dL, outpatient dietary and environmental modification should be initiated with repeat levels after 1 month. At lead levels between 45-69 mcg/dL in an otherwise well child, oral chelation therapy with dimercaptosuccinic acid (DMSA) can be started. Hospitalization with parenteral chelation therapy is indicated for lead levels ≥ 70 mcg/dL, symptoms of encephalopathy, or if DMSA is contraindicated or poorly tolerated. Main parenteral agents used together are dimercaprol (British anti-lewisite) and calcium disodium edetate (CaNa2EDTA). Both require close monitoring of renal function and electrolytes.9,10 It is important to involve the poison control center or toxicologists in the decision to initiate chelation therapy due to significant side effects of these medications. (See Table 5.)
Table 5. Chelation Agents for Lead Toxicity in Children9 |
|||
|
Lead Level |
Medication |
Dosing |
Contraindications |
|
45-69 mcg/dL |
DMSA, PO |
10 mg/kg OR 350 mg/m2 (rounded to nearest 100 mg), PO, TID x 5 days then BID x 14 days |
Ongoing exposure Hepatic dysfunction |
|
≥ 70 mcg/dL |
Dimercaprol, IM |
75 mg/m2, IM, q4hours |
Nut allergy G6PD deficiency |
|
|
CaNa2EDTA, IM or IV |
1000 mg/m2, IV infusion, daily OR 250 mg/m2, IM, q4hours |
|
|
Note m2 correlates to body surface area DMSA: dimercaptosuccinic acid; CaNa2EDTA: calcium disodium edetate; IM: intramuscular; PO: by mouth; IV: intravenous |
|||
Hemolytic Anemia
Hemolytic anemia is a hematologic emergency involving a reduction in red blood cell life span due to extrinsic forces or intrinsic red blood cell disorders. Extrinsic hemolytic anemias result from damage to red blood cells due to immunological, chemical, or physical forces. Examples of extrinsic processes include autoimmune hemolytic anemia, hypersplenism, or shear from mechanical valves causing damage to cells. Autoimmune hemolytic anemia is often preceded by upper respiratory infections.
Intrinsic hemolytic anemia can be seen in inherited abnormalities including hemoglobinopathies (sickle cell disease, thalassemias), enzyme deficiencies (glucose-6-phosphate dehydrogenase deficiency, pyruvate kinase deficiency), or membrane defects (hereditary spherocytosis, elliptocytosis). Sickle cell anemia and beta-thalassemia, hemoglobinopathies involving mutation of the beta-globin chain typically present between 6-12 months of age due to the protective predominance of fetal hemoglobin F (alpha- and gamma-globin chains only) in the neonatal period. In contrast, severe forms of alpha-thalassemia, hemoglobinopathy with mutation of alpha-globin, present at birth. Disorders involving enzyme or membrane defects manifest in the neonatal period or in early childhood in the form of acute hemolytic crises in response to medications, acute infections, or other sources of stress. For example, exposure to sulfa, nitrofurantoin, lead-chelating agents, or fava beans can lead to acute hemolytic anemia in glucose-6-phosphate dehydrogenase deficiency.
In the work-up of hemolytic anemia, family history and history of recent illnesses or medication exposures are important. Laboratory evaluation often shows elevated indirect bilirubin, increased lactate dehydrogenase, reduced haptoglobin, and > 3% reticulocytosis.3 Spherocytosis, polychromasia, schistocytes, bite cells, Heinz bodies (denatured hemoglobin precipitates), or clumped red blood cells may be seen on blood smear. The direct antiglobulin test (Coombs test), which detects antibodies and complement proteins on red blood cell surfaces, will be positive in 97-99%.11
Children with hemolytic anemia require hospitalization. Pediatric hematology should be consulted from the ED to help guide management. For autoimmune hemolytic anemia, glucocorticoids are the mainstay of treatment and are effective in 80% of cases.11,12 Intravenous methylprednisolone 1-2 mg/kg every 6 hours can be initiated if the diagnosis of autoimmune hemolytic anemia is certain. Transfusion should be avoided if possible in as autoantibodies can destroy transfused cells, leading to increased hemolysis. Other inpatient options for autoimmune hemolytic anemia include rituximab, intravenous immunoglobulin (IVIG), exchange transfusion, and splenectomy.
Sickle Cell Anemia
Sickle cell anemia is an autosomal recessive disorder affecting 1 in 500 African Americans in the United States. Adult hemoglobin is a tetramer of two alpha-globin and two beta-globin chains (α2ß2). Hemoglobin S (Hb S) results from a mutation in the beta-globin chain of hemoglobin due to a valine for glutamic acid substitution. Due to the substitution mutation, Hb S is less soluble than normal hemoglobin and tends to polymerize in deoxygenated conditions to form rigid, sickle-shaped red blood cells, which can occlude capillaries. This can occur in every organ system, especially in dehydration or stress, over time, resulting in both acute and chronic multisystem organ failure. Sickle cell anemia (Hb SS) refers to homozygosity for Hb S and manifests as the most severe form of the disease, while patients with sickle cell trait (one copy of Hb S) are asymptomatic.
In the United States, sickle cell anemia is diagnosed on newborn screening. Severe anemia and vaso-occlusive events typically do not present until around 6 months of age due to the protective predominance of fetal hemoglobin F (α2γ2) in the postnatal period. Specific syndromes highlighted in this article include acute pain crisis, acute chest syndrome, splenic sequestration, aplastic anemia, fever, and stroke. Other notable complications of sickle cell anemia include dactylitis, priapism, bone infarction, and organ infarction (commonly splenic, hepatic, or renal).
Pain Crises. In one prospective study of 3578 patients, the mean number of acute pain crises per year was 0.8 in sickle cell disease, with wide variability among individuals.13 Pain crises commonly occur in the chest, abdomen, back, and extremities. Crises can be precipitated by weather changes, exhaustion, dehydration, infection, stress, alcohol, or hypoxemia. ED evaluation includes a thorough physical exam, complete blood panel, reticulocyte count, metabolic panel, as well as radiographic studies targeted toward areas of concern.
Management of sickle cell pain crises in the ED involves a combination of supportive therapy, such as warm compresses and hydration, as well as pharmacologic therapy with nonsteroidal anti-inflammatory drugs (NSAIDs) and opiates (see Table 6). Patients may have individualized protocols based on what has been effective in previous episodes. Of note, meperidine should be avoided due to potential for central nervous system toxicity with its metabolite nor-meperidine. Re-evaluation for pain should be conducted every 15-30 minutes. Hospitalization should be considered if more than two doses of opiates are required.14
Table 6. Principles of Management of Syndromes Associated with Sickle Cell Anemia in the ED |
||
|
|
Management Principles |
Admission Criteria |
|
Pain crises |
Warm compresses, massage Gentle hydration 10 mL/kg bolus, then maintenance fluids Pain control based on previously effective regimens Options for pain control
Re-evaluate every 15-30 minutes |
Admit if more than 2 doses of opiates are required |
|
Acute chest syndrome |
Supplemental oxygen to maintain saturations > 92% Gentle hydration 10 mL/kg bolus, then maintenance fluids Analgesia as above Options for antibiotics
Transfusion for hypoxia, >10-20% drop in hematocrit, |
Admit if there is any clinical suspicion for acute chest syndrome |
|
Splenic sequestration |
Rapid recognition is critical: signs include hypovolemic shock, splenomegaly, anemia with reticulocytosis Start normal saline bolus no more than 10-20 mL/kg Initial transfusion of red blood cells at 10 mL/kg Slow rate and volume of transfusion after initial stabilization Do not exceed baseline hematocrit |
Admit to the pediatric intensive care unit |
|
Aplastic crisis |
Supportive care with IV + oral hydration at maintenance rate Transfusion gently at 5 mL/kg over 4 hours for symptomatic anemia, hypoxia, or hemodynamic instability |
Admit for observation until marrow recovery |
|
Fever |
Work-up common sources of infection including: urinary tract infection, pneumonia, osteomyelitis, meningitis, bacteremia Empiric antibiotics for all febrile children
|
Admit for observation, IV antibiotics in all high-risk groups (see list above) |
|
Stroke |
Imaging options
Exchange transfusion to reduce hemoglobin S < 30% Supportive care: IV hydration to maintain euvolemia, correct hypoglycemia and other electrolyte abnormalities |
Admit for exchange transfusion, neurology or neurosurgical consultation |
Acute Chest Syndrome. Acute chest syndrome is the second most common cause of hospitalization, and the most common cause of death in sickle cell anemia. In the ED, a high index of suspicion for acute chest syndrome is required for any child with sickle cell anemia presenting with fever, cough, dyspnea, chest pain, or hypoxia. The pathophysiology is a combination of pulmonary infarction and inflammatory response, often accompanied by superimposed infection. Chest X-ray in acute chest is often indistinguishable from pneumonia. ED management includes supportive management with supplemental oxygen, IV hydration, analgesia as in pain crises, and admission to the hospital. Antibiotics must be given to cover Streptococcus pneumoniae, Hemophilus influenzae, and Mycoplasma pneumoniae. Transfusion or exchange transfusion is indicated for hypoxia, anemia with hematocrit 10-20% below patient’s baseline, multi-lobar infiltrates, or clinical deterioration.14,15 Observational studies show that exchange transfusion, which involves removing a patient’s blood and transfusing allogeneic blood with the goal of dilution the concentration of Hb S, may be superior to simple blood transfusion in acute chest.15
Splenic Sequestration. Splenic sequestration is a life-threatening complication of sickle cell anemia with 10-15% mortality rate. It typically occurs in young children ages 6 months to 2 years prior to complete splenic infarction. Sequestration occurs when vaso-occlusion in the spleen results in trapping of red blood cells within splenic sinusoids. Children present with left upper quadrant pain, massive splenomegaly, and acute drop in hemoglobin (can be as low as 1-3 g/dL) with reticulocytosis. Rapid recognition is critical, as these patients can progress rapidly to hypovolemic shock. ED management can begin with intravascular support with normal saline, followed by red blood cell transfusion (see Table 6). Hematocrit should not be transfused above baseline due to risk of hyperviscosity as the spleen contracts and auto-transfusion occurs. Definitive management is splenectomy after any life-threatening event as recurrence can be as high as 50%.16
Aplastic Crisis. Aplastic crisis is marked by transient episodes of declining hemoglobin concentration in the setting of reticulocytopenia. Episodes are often preceded by infection (commonly Parvovirus B19), with spontaneous recovery occurring around 1 week. In contrast to splenic sequestration, patients rarely present with hypovolemic shock. This is because marrow suppression in aplastic anemia progresses over the course of several days, allowing for compensatory mechanisms such as increased blood volume or cardiac output to develop. Patients with aplastic crisis should be admitted to the hospital for observation until marrow recovery. Given the possibility of high output heart failure, fluids or transfusion should be given slowly and cautiously if indicated.15,16 (See Table 6.)
Fever. Infection is the most common cause of death in children with sickle cell disease. Therefore, fever in sickle cell patients warrants emergent evaluation. Functional asplenia begins to occur around 3 months of age and leads to susceptibility to bacterial infections, in particular, by S. pneumoniae, H. influenzae, Neisseria meningitides, and M. pneumoniae.14,17 As outpatients, these children should receive vaccination against pneumococcus at 6 months and 2 years of age, as well as every 5 years after. Prophylactic penicillin should be started at 2 months.
In the ED, workup for fever in the sickle cell patient includes a thorough physical exam, CBC with differential, reticulocyte count, blood cultures, urinalysis and culture, and appropriate radiographic studies. Chest X-ray is indicated if there is cough, chest pain, tachypnea, or hypoxia. Any painful, erythematous, or swollen extremity should be imaged to evaluate for osteomyelitis; Staphylococcus aureus and Salmonella are the most common causative organisms. Lumbar puncture should be performed in children with altered mental status, seizure, or meningismus to evaluate for meningitis.
Empiric coverage with a broad-spectrum antibiotic with good blood-brain barrier penetration (e.g., ceftriaxone) is indicated in all febrile children with sickle cell anemia. Vancomycin should be added if there is a history of Methicillin-resistant S. aureus (MRSA), high prevalence of MRSA in the community, or if there is concern for meningitis. Inpatient admission is strongly recommended in the following groups:14,17
-
Any child who is ill-appearing or has poor oral intake
-
Children ≤ 12 months old
-
High fever ≥ 40º C
-
History of sepsis, meningitis, or recent antibiotic use
-
Hemoglobin levels lower than baseline
-
Hypoxia or infiltrate on chest X-ray
-
Inability to obtain next-day follow-up
Stroke. Stroke is a major cause of morbidity and mortality in children with sickle cell anemia. The prevalence of stroke in sickle cell anemia is 11% in patients younger than 20 years of age, with the peak incidence occurring between 2-5 years of age.18 Sickle cell patients are predisposed to both ischemic and hemorrhagic stroke. Ischemic strokes may present with sensory or motor deficits, hemiparesis, aphasia, or seizure. Strokes involving the posterior circulation may present with ataxia, vertigo, or vomiting. Any new or sudden-onset headache warrants suspicion for hemorrhage. Silent infarcts without overt symptoms are seen in 21.8% of children between 6-19 years old.
Work-up in the ED involves a thorough neurologic exam, labs (e.g., blood glucose, CBC, reticulocyte count), and imaging. Non-contrast CT scan of the head may be indicated if there is concern for hemorrhage. Otherwise, magnetic resonance imaging with angiography and venography (MRI/MRA with venography) is the preferred imaging modality, as it can evaluate for vasculopathy as well as dural venous thrombosis. Treatment is immediate exchange transfusion, with goal reduction of Hb S < 30%. Neurology or neurosurgical (if hemorrhagic stroke) consultation should be obtained.
Neonatal Polycythemia
Polycythemia, defined by a peripheral blood venous hematocrit or hemoglobin level > 2 standard deviations above normal, is seen in 1-5% of newborns. For term infants, this is equivalent to a hematocrit > 65% or hemoglobin > 22 g/dL. The most common cause is delayed clamping of the umbilical cord, resulting in increased placental blood transfusion to the patient. Other causes include increased erythropoiesis in the setting of chronic intrauterine hypoxia, which can be seen in the setting of preeclampsia or other maternal cardiac or pulmonary disorders.
Symptoms, if they do arise, often occur within hours of birth. Thus, if patients are asymptomatic by day 2-3 they are likely to remain asymptomatic. Polycythemic newborns should be closely observed for symptoms of cyanosis, tachypnea, and poor feeding. Oral intake, body weight, and urine output should be monitored for hydration status. Partial exchange transfusion can be considered for patients who are symptomatic or when the hematocrit is > 70%. However, multiple studies show that partial exchange transfusion may not confer long-term neurodevelopmental benefits, and that crystalloids may be as effective.19
Thrombocytopenia
Platelets are derived from megakaryocytes from the myeloid stem cell lineage, have a lifespan of 8-10 days, and are important in clot formation. Thrombocytopenia is defined as a platelet count of < 150,000/uL. The risk of bleeding is increased when the platelet count is < 100,000/uL, and the risk of spontaneous bleeding is high when platelets are < 20,000/uL. As in anemia, it is important to first rule out blood loss, as hemorrhage leads to thrombocytopenia. Other etiologies of thrombocytopenia can be categorized into disorders of platelet production and that of increased platelet destruction (see Table 1). Examples of platelet destruction include immune thrombocytopenia (ITP) and hemolytic-uremic syndrome (HUS), which will be discussed further in this review. Congenital disorders in platelet function, such as Wiskott-Aldrich or Glanzmann thrombasthenia, will not be discussed in this review.
The History and Physical Exam in Thrombocytopenia
Thrombocytopenia is often diagnosed incidentally on routine blood tests or in the work-up for easy bleeding or bruising. In the hemodynamically unstable patient, ED priorities include stabilizing airway, breathing, and circulation. In the stable patient, workup of thrombocytopenia starts with a detailed history of easy bleeding or bruising, mucosal hemorrhages, menorrhagia, or systemic symptoms of fever, bone pain, or weight loss. Preceding factors are also important. For example, ITP is typically preceded by a viral infection, while HUS is preceded by gastrointestinal symptoms. Medication review is also an important part of the history, as thrombocytopenia can be a side effect of many medications (e.g., antipyretics, antibiotics, anticonvulsants) that resolves with removal of the offending agent. Notable exam findings in thrombocytopenia include mucosal bleeding (e.g., epistaxis, gingival bleeds), petechiae, or purpura. In contrast to hemophilia (factor 8 or 9 deficiency), thrombocytopenia tends to present with prolonged bleeding after superficial injury instead of bleeding into deep tissues or joints.
Immune Thrombocytopenia
ITP is a common and usually benign condition of childhood, characterized by isolated low platelet count < 100,000/uL. Primary ITP in children involves autoantibodies directed against antigens in the platelet membrane, which targets platelets for destruction by the reticuloendothelial system. In 60% of cases, there is a history of preceding viral infection. Pediatric ITP is clinically distinct from adult ITP due to a lower incidence of associated comorbidities and a higher likelihood of spontaneous remission. Prognosis in children is favorable, as 50% recover in 1-2 months, 75% by 6 months. 20% have chronic ITP, defined as thrombocytopenia lasting more than 12 months.
Clinical manifestations include petechial rash, purpura, and mucosal or other bleeding occurring in an otherwise well-appearing child. There should be no systemic complaints of fever, anorexia, malaise, weight loss, bone pain, abdominal pain, lymphadenopathy, or significant hepato/splenomegaly. According to a database of 1784 children with ITP, 80% presented with platelet counts as low as 20,000/uL and 44% with platelets < 10,000/uL.20 Aside from thrombocytopenia, the hemogram should be normal, without significant neutropenia, anemia, and without schistocytes or other signs of hemolysis on blood smear.
Most cases of ITP can be discharged from the ED without pharmacological intervention as long as follow-up can be arranged. Parents should be counseled regarding restricting contact sports or activities that increase the risk of trauma, helmet use in the appropriate setting, and avoiding antiplatelet medications such as NSAIDs. Patients will need periodic platelet monitoring as an outpatient starting at weekly intervals for at least 3 months.
Pharmacologic intervention is indicated in life-threatening bleeding, which occurs in 3% of cases, or those with severely impaired quality of life. Examples include severe epistaxis, gastrointestinal bleeding, and intracranial hemorrhage (ICH). In a study of 120 children with ITP, 75% of those with ICH had platelet counts < 10,000/uL compared with 59% in matched controls. Those with ICH were also more likely to have had head trauma.21
Steroids and IVIG are the main pharmacologic options in the treatment for ITP (see Table 7). There is some evidence that IVIG may result in faster recovery of platelets within 48 hours compared with steroids.22,23 Anti-D immunoglobulin may be considered in patients who are Rh-positive, with a negative direct antiglobulin test, and have not had splenectomy. Platelet transfusion is contraindicated unless there is life-threatening hemorrhage. Pediatric hematology should be consulted prior to starting IVIG, steroids, or transfusion.
Table 7. Pharmacologic Treatment Options for Immune Thrombocytopenia22 |
|
|
Corticosteroids |
Oral prednisone 2-4 mg/kg/day (max 240 mg/day) Oral prednisone 2-4 mg/kg/day (max 240 mg/day) IV methylprednisolone 30 mg/kg (max 1 g/day) *Continue until platelet count improves, usually 3-5 days. Longer steroid courses will need a taper over 3-4 weeks |
|
IVIG |
0.8-1.0 g/kg x 1 dose |
|
Anti-D |
75 mcg/kg x 1 dose |
Hemolytic Uremic Syndrome
HUS is a hematologic emergency defined by a triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. It mainly affects children younger than 5 years of age. HUS is a toxin-mediated phenomenon with Shiga toxin producing Escherichia coli 0157:H7 (STEC), accounting for 70-90% of cases in the United States. Briefly, Shiga-toxin in the gastrointestinal mucosa can translocate to the bloodstream where it binds to erythrocytes, platelets, and monocytes eliciting an inflammatory cascade that leads to HUS.24 Other associated organisms include Pneumococcus (5-15% cases in the United States) and Shigella. The use of antibiotics and anti-motility medications during diarrheal illness may increase the risk of HUS.25
The onset of HUS is typically 5-10 days after a gastrointestinal illness consisting of abdominal pain, vomiting, and bloody diarrhea. Children often present to the ED acutely ill with pallor, oliguria, and other signs of multi-organ dysfunction. Hypertension, pulmonary edema, and other signs of volume overload may be seen as a result of kidney failure. CNS manifestations include seizure, coma, or stroke. Gastrointestinal manifestations include hemorrhagic colitis, bowel necrosis, peritonitis, and bowel perforation.
Work-up for HUS should be initiated in children with any of the above findings on history or exam. CBC and basic metabolic in HUS will show creatinine elevation, anemia with hemoglobin < 8/dL, and thrombocytopenia with platelets < 140,000 /uL (often < 40,000 /uL). Hemolysis with abundant schistocytes and helmet cells due to shearing of red cells by platelet microthrombi can be seen on blood smear. Coagulation studies such as prothrombin time, partial thromboplastin time, and fibrinogen levels should also be obtained to distinguish HUS from disseminated intravascular coagulation. Testing for shiga-toxin in the stool or serum (PCR or ELISA) can be sent as well.
All children with HUS or suspected HUS should be admitted to the hospital. Pediatric hematology should be consulted to guide management, which is largely supportive. In the ED, fluid and electrolyte balance need to be monitored closely. Dialysis is necessary in 50-60% of cases, and is indicated in symptomatic uremia, azotemia (BUN > 80 mg/dL), severe volume overload, or refractory electrolyte abnormality. Blood products should be avoided if possible, as transfusion may worsen hemolysis and renal failure. If transfusion is necessary, as in cases of hemodynamic instability or bleeding, blood products should be leukoreduced in case of future need for renal transplantation. For severe hypertension or signs of hypertensive emergency (e.g., seizure, stroke, altered mental status), calcium channel blockers (nifedipine or nicardipine) may be used. Prognosis is generally favorable with mortality < 5%, with most children experiencing complete resolution within 1-2 weeks. Approximately 5% have long-term complications such as neurological dysfunction or end-stage renal disease.26
Leukocyte Disorders
Neutrophils, which typically comprise 60-70% of circulating leukocytes, are of particular significance as they are responsible for immunity against bacterial and fungal infections.1 Elevated neutrophils can be seen in acute infection as well as any state of acute inflammation or stress. Immature neutrophils, known as bands, typically comprise < 10% circulating leukocytes. Studies show bandemia, especially at levels > 20%, can be indicative of bacteremia.27 However, other studies show specificity of bandemia for severe infection is poor, limiting its clinical utility.28
Another situation in which leukocytosis is encountered in the ED is the new diagnosis of leukemia or lymphoma. Neutropenia can be seen as a complication of chemotherapy. These pediatric oncology patients tend to be challenging to the ED physician due to complex medical histories, lack of familiarity with chemotherapeutic regimens, or absence of easily accessible consultant specialties in the community setting. The workup and management of two emergent oncologic conditions — neutropenic fever and leukostasis — will be discussed in this section.
The History and Physical Exam in the Evaluation of Leukocyte Disorders
As in anemia and thrombocytopenia, assessment of hemodynamic stability is the first priority of the ED physician in the evaluation of leukocyte disorders. In the stable patient with undifferentiated neutropenia, family history is important. For example, cyclic neutropenia is a rare, autosomal dominant disorder resulting in neutropenia every 2-4 weeks starting in the first year of life. Due to intact monocyte and lymphocyte-mediated immunity, these children are not as high risk for infection as post-chemo patients.29 Medications such as clozapine and sulfasalazine can also cause neutropenia, as can hepatitis, HIV, and EBV infection. In both leukocytosis and leukopenia, screening for sources of infection, such as upper respiratory, urinary symptoms, and gastrointestinal symptoms, is essential. A thorough physical should also include the nail, skin, perineal, and oropharyngeal exam.
Neutropenic Fever in the Chemotherapy Patient
Febrile neutropenia is a frequent complication of chemotherapy in pediatric cancer patients. Neutropenia is defined as absolute neutrophil count (ANC) < 1500 cells/uL. In the pediatric cancer patient, significant neutropenia is defined as ANC < 500 cells/uL or expected to decrease to that level within 48 hours. In this population, fever is defined as a single oral temperature of ≥ 38.3º C (101º F), or temperature > 38.0º C (100.4º F) sustained more than 1 hour or more than two elevations within a 12-hour period.
Evaluation in the ED should begin with a thorough history and physical exam focusing on potential sources of infection. Important history includes antimicrobial prophylaxis, previous infections, timing of last chemotherapy, and presence of intravascular catheters or other implanted devices. Physical exam should pay special attention to the skin, perineum, oral mucosa, and central venous line sites. ED studies include complete blood count with differential and blood cultures, including samples from all lumens of central venous catheters, urinalysis and culture, and chest X-ray if there are respiratory complaints. No invasive procedures, such as urinary catheterization or rectal examination, should be performed.
Of note, documented infection in febrile neutropenia is found in 25-50% of cases, while no clear source is found in more than 50%.30,31 The most common documented infection is bacteremia. Frequently isolated organisms include coagulase-negative Staphylococcus, Streptococci viridans, S. aureus, MRSA, E. coli, Pseudomonas spp., and Klebsiella sp.
Although some studies show outpatient management with oral antibiotics (often fluoroquinolone monotherapy, or fluoroquinolone and amoxicillin-clavulanate) may be safe in low-risk groups, no single risk prediction rule has been validated. As such, most institutions support inpatient hospitalization and empiric IV antibiosis for all children with neutropenic fever, with discontinuation of antibiotics if blood cultures are negative at 48 hours and patients are afebrile for at least 24 hours.32 Initial antibiotic choice in the ED should take into account the patient’s likely source of infection, prior history or culture, and community resistance profiles. Monotherapy with a broad-spectrum antibiotic with pseudomonas coverage is generally adequate (see Table 8). Vancomycin should be added if there is hypotension or hemodynamic instability, radiographic evidence of pneumonia, skin or soft tissue infection, or suspected central line infection.
Table 8. Antibiotic Treatment Options for Pediatric Febrile Neutropenia |
|
|
Empiric Monotherapy Options |
|
|
Cefepime |
50 mg/kg IV every 8 hours (max 2 g per dose) OR |
|
Ceftazidime |
50 mg/kg IV every 8 hours (max 2 g per dose) OR |
|
Meropenem |
≥ 3 months old, 20 mg/kg IV every 8 hours |
|
Piperacillin/tazobactam |
< 9 months, 80 mg/kg IV every 8 hours ≥ 9 months and < 40 kg, 100 mg/kg IV every 8 hours ≥ 40 kg, 3 g IV every 6 hours or 4 g every 6-8 hours (max 16 g/day) |
|
PLUS vancomycin if indicated* |
|
|
Vancomycin |
15 mg/kg IV every 6 hours |
|
Dosages will need to be adjusted for renal dysfunction *Vancomycin is indicated for hypotension, hemodynamic instability, radiographic evidence of pneumonia, skin or soft tissue infection, or suspected central line infection. |
|
Neutropenic Fever in Non-chemotherapy Patients
As mentioned previously, neutropenia can be seen in non-oncologic processes as well, such as in cyclic neutropenia, post-viral myelosuppression, or as a side effect of medications. Patients with chronic benign neutropenia and cyclic neutropenia should be admitted for parenteral antibiotics if they are ill-appearing or have a history of complicated febrile illness. Often in the ED, the underlying etiology is unknown. These children with undifferentiated febrile neutropenia should receive evaluation and antibiotics similar to chemo-related neutropenic fever, as well as admission for a thorough oncologic and infectious workup.
Leukostasis
Hyperleukocytosis is defined as total white blood cell count ≥ 100,000/uL. In the pediatric setting, hyperleukocytosis can be seen in the initial presentation of acute lymphoblastic leukemia and acute myeloid leukemia. Leukostasis refers to hyperleukocytosis with symptoms of decreased tissue perfusion. White blood cell levels ≥ 300,000/uL are especially at high risk of symptomatology. The pathophysiology behind leukocytosis involves increased blood viscosity in addition to blast cells, which tend to be more rigid than mature leukocytes, leading to capillary occlusion and eventually tissue/organ ischemia.34
Microvascular occlusion in the central nervous system and pulmonary system are the major causes of mortality in leukostasis. Patients can present with headache, somnolence, altered mental status, or coma. Hemiplegia or focal neurologic deficits can be seen as a result of stroke or ICH. Pulmonary infarction and infection can occur. Eighty percent of patients are febrile on presentation due to inflammatory response, in which case blood cultures and antibiotic should be started empirically. Other complications of leukostasis include cardiac ischemia, renal insufficiency, bowel infarction, priapism, or limb ischemia.
Laboratory tests in the ED include complete blood count with blood smear. It is also critical to monitor electrolytes for concomitant tumor lysis syndrome (TLS), marked by hyperkalemia, hyperphosphatemia, hyperuricemia, and hypocalcemia. Although TLS is often associated with initiation of chemotherapy, it can be seen in the initial presentation of leukemia as well. The most important treatment the ED physician can offer in leukostasis is aggressive IV hydration starting with a 10-20 mL/kg bolus, followed by crystalloids at 1.5-2 times the maintenance rate. Definitive treatment is induction chemotherapy, which can reduce leukemic count within 24 hours. For patients in whom induction chemotherapy must be delayed, such as those with TLS, alternative treatment with hydroxyurea or leukopheresis may be discussed with the oncology consulting team. The initial mortality rate of leukostasis can be high as 5% in acute lymphoblastic leukemia, and 20-40% in acute myeloid leukemia in the acute period.34,35
Summary
Children and adolescents with hematologic abnormalities can be some of the most complicated patients presenting to the ED. The spectrum of disease can range from relatively benign conditions, such as iron deficiency anemia and immune thrombocytopenia, to life-threatening diseases, such as hemolytic anemia and hemolytic uremic syndrome. Leukocyte disorders, such as neutropenia or leukocytosis, can be seen in pediatric cancer patients who often have complex medical histories. In most cases, a focused history and a thorough physical exam are critical to guide the physician toward subsequent work-up and eventual diagnosis. The ED physician must be facile with interpretation of laboratory results and key management principles for common and life-threatening hematologic conditions.
REFERENCES
- Wickramasignhe SN, Erber WN. Chapter 1: Normal blood cells. Blood and Bone Marrow Pathology. 2nd ed. Edinburgh: Elselvier; 2011.
- Nathan DG, Oski FA. Nathan and Oski’s Hematology of Infancy and Childhood. 6th ed. Philadelphia, PA: WB Saunders; 2003:1841.
- Sadowitz PD, Amanullah S, Souid AK. Hematologic emergencies in the pediatric emergency room. Emerg Med Clin North Am 2002;20:177-198.
- Centers for Disease Control and Prevention (CDC). Iron deficiency — United States, 1999-2000. MMWR Morb Mortal Wkly Rep 2002;51:897-899.
- Brotanek JM, Gosz J, Weitzman M, Flores G. Iron deficiency in early childhood in the United States: Risk factors and racial/ethnic disparities. Pediatrics 2007;120:568-575.
- McMillan JA, Oski FA, Lourie G, et al. Iron absorption from human milk, simulated human milk, and proprietary formulas. Pediatrics 1977; 60:896-900.
- Baker RD, Greer FR, Committee on Nutrition American Academy of Pediatrics. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0-3 years of age). Pediatrics 2010;126:1040-1050.
- Centers for Disease Control and Prevention (CDC). Blood lead levels — United States, 1999-2002. MMWR Morb Mortal Wkly Rep 2005;54:513-516.
- Woolf AD, Goldman R, Bellinger DC. Update on the clinical management of childhood lead poisoning. Pediatr Clin North Am 2007;54:271-294.
- American Academy of Pediatrics Committee on Environmental Health. Lead exposure in children: Prevention, detection, and management. Pediatrics 2005;116:1036-1046.
- Teachey DT, Lambert MP. Diagnosis and management of autoimmune cytopenias in childhood. Pediatr Clin North Am 2013;60:1489-1511.
- Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: An update. Expert Rev Hematol 2011;4:607-618.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11-16.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033-1048.
- Josephson CD, Su LL, Hillyer KL, Hillyer CD. Transfusion in the patient with sickle cell disease: A critical review of the literature and transfusion guidelines. Transfus Med Rev 2007;21:118-133.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: The sick(led) spleen. Br J Haematology 2014;166:165-176.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762-769.
- Webb J, Kwiatkowski JL. Stroke in patients with sickle cell disease. Expert Rev Hematol 2013;6:301-316.
- Sarkar S, Rosenkrantz TS. Neonatal polycythemia and hyperviscosity. Semin Fetal Neonatal Med 2008;13:248-255.
- Kühne T, Berchtold W, Michaels LA, et al. Newly diagnosed immune thrombocytopenia in children and adults: A comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica 2011;96:1831-1837.
- Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): Study of 40 cases. Blood 27009;114:4777-4783.
- Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190-4207.
- Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: A systematic review and meta-analysis of randomized controlled trials. J Pediatr 2005;147:521-527.
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005;16:1035-1050.
- Bell BP, Griffin PM, Lozano P, et al. Predictors of hemolytic uremic syndrome in children during a large outbreak of Escherichia coli O157:H7 infections. Pediatrics 1997;100:E12.
- Siegler RL, Milligan MK, Burningham TH, et al. Long-term outcome and prognostic indicators in the hemolytic-uremic syndrome. J Pediatr 1991;118:195-200.
- Drees M, Kanapathippillai N, Zubrow MT. Bandemia with normal white blood cell counts associated with infection. Am J Med 2012;125:1124.e9-1124.e15.
- Tran Q, Place R. Severe bandemia is not a strong predictor of serious bacterial illness in emergency department pediatric patients. Ann Emerg Med 2012;60:S3.
- Souid AK. Congenital cyclic neutropenia. Clin Pediatr 1995;34:151-155.
- Hakim H, Flynn PM, Knapp KM, et al. Etiology and clinical course of febrile neutropenia in children with cancer. J Pediatr Hematol Oncol 2009;31:623-629.
- Agyeman P, Aebi C, Hirt A, et al. Predicting bacteremia in children with cancer and fever in chemotherapy-induced neutropenia: Results of the prospective multicenter SPOG 2003 FN study. Pediatr Infect Dis J 2011;30:e114-e119.
- Lehrnbecher T, Phillips R, Alexander S, et al. Guidelines for the management of fever and neutropenia in children with cancer and/or undergoing hematopoietic stem-cell transplantation. J Clin Oncol 2012;30:4427-4438.
- Rosa RG, Goldani LZ. Cohort study of the impact of time to antibiotic administration on mortality in patients with febrile neutropenia. Antimicrob Agents Chemother 2014;58:3799-3803.
- Porcu P, Cripe LD, Ng EW, et al. Hyperleukocytic leukemias and leukostasis: A review of pathophysiology, clinical presentation and management. Leuk Lymphoma 2000;39:1-18.
- Eguiguren JM, Schell MJ, Crist WM, et al. Complications and outcome in childhood acute lymphoblastic leukemia with hyperleukocytosis. Blood 1992;79:871-875.
MONOGRAPH: In-depth disorders of the hemogram: red blood cells, platelets, and white blood cells.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.