Four Thrombocytopenic Emergencies
Thrombocytopenia is encountered commonly in the emergency department (ED). A wide array of conditions and disorders can cause thrombocytopenia. In most instances, the emergency physician will not be able to determine the definitive diagnosis, but it is important that the initial evaluation be started in a timely manner and that appropriate specialists be consulted from the ED.
This article will review four emergent hematologic conditions associated with thrombocytopenia: thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), disseminated intravascular coagulation (DIC), and immune thrombocytopenia (ITP). During the last 20 years, there has been advancement in our understanding of the pathophysiology of these conditions and some improvement in the treatment. It has been determined that TTP and HUS are two separate disease entities, and not a spectrum of one condition. DIC usually is found in hospitalized patients with serious infections, although some patients may have manifestations during time in the ED. While many novel therapies have been proposed to treat DIC, none have proven beneficial. The classification of ITP has been changed extensively over the past few years, and there are many new therapies that are challenging the standard treatment of steroids and splenectomy. This article will discuss the general approach to identification and initial treatment for each of these conditions.
— The Editor
Thrombotic Thrombocytopenic Purpura
Introduction
TTP is a rare, life-threatening hematologic disorder. It was described first in 1924 as a set of clinical criteria including fever, thrombocytopenia, microangiopathic hemolytic anemia, acute kidney injury, and neurologic symptoms. TTP historically has a mortality nearing 90% if left untreated.1,2 Given the high mortality and unknown etiology, it became an area of increasing study in hematology.
Current understanding of this disorder has evolved beyond the five features noted above. The pathophysiology of TTP now is understood to involve a deficiency in activity of ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). ADAMTS13 is an ezyme responsible for cleaving large von Willebrand factor (vWf) polymers, thus ADAMTS13 also is known as von Willebrand factor-cleaving protease. In TTP, ADAMTS13 activity is usually less than 10% of normal. This leads to the accumulation of large vWF polymers and resultant microthrombi that cause end organ damage. As a result, more recent definitions of TTP include deficiency in ADAMTS13 activity along with the characteristic clinical features.3 As the understanding of the disease evolved, plasma exchange was recognized as a useful treatment, decreasing mortality to 10%.1,2
Previously, TTP was considered to be on a spectrum of disease with HUS, as both fall into the category of thrombotic microangiopathies (TMA), a group of disorders identified as causing thrombocytopenia, hemolytic anemia, and end organ damage as a result of microvascular thrombi. However, as further understanding of the etiology, pathophysiology, and treatment of HUS and TTP has evolved, combining these diseases into a single spectrum has been discouraged.
Epidemiology
Data from TTP registries allow for estimates of its incidence, which ranges from 3 to 13 cases per million persons per year.4,5 The majority of these cases, nearly 90%, are reported in adults.5,6 Only nine pediatric cases per 100 million children per year occur.5 Risk factors for development of TTP include female sex and African-American race.5,7
Etiology
TTP was described first by the following set of criteria: fever, thrombocytopenia, microangiopathic hemolytic anemia, acute kidney injury, and neurologic signs. It was diagnosed clinically in the absence of any other obvious cause of these criteria. Over time, however, the understanding of the underlying etiology changed. Large vWF multimers were observed in patients with recurrent TTP and ultimately were implicated in the pathophysiology of the disease. ADAMTS13, a protease responsible for cleaving large vWF multimers, subsequently was isolated. A deficiency in ADAMTS13 then was found to play a critical role in development of large vWF mulitmers and TTP.8,9 In as many as 94% of cases, ADAMTS13 deficiency results from the development of autoantibodies to ADAMTS13.4 In the remaining cases, TTP can be an inherited entity resulting from a decrease in the ADAMTS13 protein.
Pathophysiology
The underlying pathophysiology of TTP revolves around the deficiency in ADAMTS13, whether it is inherited or acquired, and the development of large vWF polymers. Under normal physiologic conditions, vWF is secreted from healthy endothelial cells and circulates in multimers.10 vWF binds to damaged endothelial cells and platelets to promote thrombus formation in damaged vessels. ADAMTS13 cleavage sites are exposed on vWF when circulating multimers become too large, allowing for cleavage into smaller sized multimers.3,10 In the absence or deficiency of ADAMTS13, vWF multimers become progressively larger. This leads to progressive platelet binding and the formation of microthrombi that occlude microvasculature, leading to end organ damage, such as acute kidney injury and neurologic symptoms. Formation of the microthrombi consumes platelets and creates turbulent blood flow in the microvasculature. The turbulent blood flow is thought to cause shear force on passing red blood cells.10 This leads to damage, formation of schistocytes, and resultant hemolytic anemia.
Presentation and Diagnosis
Patients have a variety of presentations, with the traditional pentad present at diagnosis in only 5% of cases.11 Most commonly, in 69% of cases, patients present with gastrointestinal (GI) symptoms including abdominal pain, nausea, and vomiting. An additional 66% have neurologic symptoms, ranging from headache to altered mental status, seizure, and focal deficit.11 Fever and chest pain are present at diagnosis in 23% and 22%, respectively.11
The hallmark of diagnosis of TTP is evidence of a thrombotic microangiopathy, manifested as thrombocytopenia with hemolytic anemia and evidence of schistocytes. These findings should prompt measurement of ADAMTS13 activity.11,12 Given the broad differential and variety of patient presentations, attention must be paid to exclude other causes of the patient’s illness.
Diagnosis of TTP in the ED often is made on a presumptive basis. Presumptive diagnosis should be made if the patient is found to have microangiopathic hemolytic anemia (often seen as schistocytes) and thrombocytopenia without another apparent cause.11 Fever, acute kidney injury, and neurologic symptoms need not be present for a presumptive diagnosis. Given the low incidence of the disease, a high degree of suspicion must be maintained when evaluating patients. Additionally, care must be taken to evaluate for other disorders that may present similarly. A differential diagnosis is listed in Table 1. An initial approach can be to rule out infection, DIC, and other immediately life-threatening etiologies. An initial workup may include complete blood count (CBC), chemistry, LFT, PT/INR, fibrinogen, blood cultures, chest X-ray, urinalysis, lactate, as well as any other studies tailored to patient presentation and complaint. One differentiating feature of TTP and DIC is that coagulation studies tend to be normal in TTP.
However, once TTP is suspected, further discussion with a hematologist should occur, given the need for plasma exchange. Usually, therapy is initiated empirically in the ED before the ADAMTS13 result is available. ADAMTS13 activity generally is used to guide further therapy.
Table 1. Differential Diagnosis for Thrombotic Thrombocytopenic Purpura
- Hemolytic uremic syndrome
- Transient ischemic attack
- Malignancy
- Disseminated intravascular coagulation
- Sepsis
- Vasculitis
- Immune thrombocytopenia
- Human immunodeficiency virus
- Heparin-induced thrombocytopenia
- Cerebrovascular accident
- Systemic lupus erythematosus
Management
Initial management in the ED relies first on recognition of the rare disease and a workup to evaluate for other etiologies of the patient’s presentation. Early recognition is essential, as TTP has a mortality rate nearing 90% if left untreated.1,2 Even if recognized and managed appropriately, the mortality rate remains at 10-20%.1,2
Once the diagnosis is suspected and presumptive diagnosis made, the primary treatment modality is plasma exchange.6,11-14 This may be initiated prior to ADAMTS13 testing. Once plasma exchange is initiated, it continues until platelets recover and hemolysis ceases.6 Use of plasma exchange is responsible for a marked improvement in mortality and is a Class 1, Grade 1A recommendation in TTP by the American Society for Apheresis.13 Plasma exchange is a specialized treatment, and discussion with a hematologist is necessary for proper performance. Furthermore, if plasma exchange is unavailable, the patient should be transferred to a higher level of care, possibly to a tertiary care center where hematology is more readily available.
After early recognition and plasma exchange, the remainder of care is mainly supportive. This includes intravenous (IV) fluids as needed, vasopressor agents for refractory hypotension, intubation for respiratory failure, and transfusion of packed red blood cells as needed for anemia.
Conversely, transfusion of platelets to correct the thrombocytopenia of TTP remains controversial, as there are reports of deterioration after platelet transfusion in TTP.15,16 This led to the thought that platelets may worsen TTP disease by increasing thrombosis, conceptualized as an “adding fuel to the fire” model. Recent data from the Oklahoma registry suggest that this may have been overstated, as 61% of patients in this registry had received platelets prior to diagnosis with no increase in mortality or neurologic outcomes.17 The exact role of platelet transfusion in TTP remains controversial. Many sources recommend against transfusion unless the patient is experiencing life-threatening bleeding or requires an invasive procedure.
Summary
TTP is a clinically distinct disease with hallmarks of thrombocytopenia, hemolytic anemia, and deficiency in ADMATS13. It is a rare entity with high mortality if left untreated, thus necessitating a high degree of suspicion in patients presenting with characteristic features. Once the disease is suspected, initiation of plasma exchange along with supportive care, discussions with a hematologist and transfer to an appropriate care center are of proven benefit.
Hemolytic Uremic Syndrome
Introduction
HUS was recognized initially in 1955, with a subsequent description of its cardinal features in 1962.18,19 It is a clinically distinct TMA characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. HUS usually develops following an acute diarrheal illness, most commonly Escherichia coli, although it can develop without the inciting infection. With a median age of onset of 4 years, HUS is primarily a disease of childhood.20 HUS continues to remain clinically important today, as it is one of the leading causes of acute renal failure in pediatrics.21
It is now known that HUS represents several overlapping disorders: Shiga toxin-HUS (ST-HUS), atypical HUS (aHUS), and secondary HUS. ST-HUS, by far the most common subtype, follows an acute diarrheal infection caused by Shiga toxins. aHUS is caused by dysfunction of the complement system. Secondary HUS results from coexisting illness or insult, which can include malignancy, infection, drugs, transplant, or autoimmune disorders. Each variant of HUS has its own pathophysiology and slight differences in treatment; however, all result in a common pathway leading to the clinical syndrome of HUS.
Epidemiology
Of the three HUS variants, ST-HUS accounts for 90% of all cases, with aHUS making up most of the remaining 10%.22,23 Secondary HUS represents a minimal portion of disease burden.
HUS is primarily a disease of early childhood, and 75% of cases occur by age 6 years.20,22 Incidence in the United States has been found to be 0.78 cases per 100,000 children, although incidence and prevalence vary based on region. Variance in incidence commonly is attributed to food-handling practices and presence of cattle, as ST-HUS often is precipitated by food and water contaminated by E. coli. Incidence varies worldwide, but the authors of one study in Italy found similar incidence to that in the United States.22 HUS has been shown to have a predilection for the female sex and occurs more commonly in the summer months.20,23
Etiology
HUS has three distinct subtypes, each with its own etiology and pathophysiology. ST-HUS occurs as a result of infection by Shiga toxin-producing organisms. Shiga toxin was isolated initially in Shigella dysenteriae Type 1, but E. coli O157:H7 is the most common etiologic agent in the United States.24 Typically, patients with HUS consume the bacteria through contaminated food and water, which leads to infection. Roughly 6% of patients with Shiga toxin-producing E. coli infections will go on to develop ST-HUS.23
While ST-HUS is post-infectious, the etiology of aHUS stems from dysregulation of the complement system. Mutations in the complement system account for 60% of all aHUS cases.25 Complement factor H (CFH), a regulatory protein, is affected most commonly and is the etiology in 25% of aHUS cases.26 Complement factor I, complement factor B, membrane cofactor protein, and complement factor 3 make up an additional 30% of mutations.26 The remaining aHUS cases are caused by additional complement proteins, non-complement proteins, and autoantibodies to the complement pathway.
Secondary HUS occurs as a result of another ongoing disease process and makes up only a marginal amount of all HUS cases. As a result, its etiology is not as well understood. Autoimmune disorders, malignancy, cytotoxic agents, infection, and organ transplant all have been implicated in cases. (See Table 2.)
Table 2. Etiology and Pathophysiology of Hemolytic Uremic Syndrome Subtypes
Class |
Etiology |
Pathophysiology |
|
Shiga toxin hemolytic uremic syndrome |
Shiga toxin |
Toxic effect on cells leads to apoptosis, inflammation, and platelet activation |
|
Atypical hemolytic uremic syndrome |
Deficiency in complement pathway |
Complement pathway activation leads to direct cellular damage and platelet activation |
|
Secondary hemolytic uremic syndrome |
Medication, malignancy, pregnancy, infection |
Varies by etiology |
Pathophysiology
ST-HUS begins with infection of the host agent, most commonly by E. coli, which is a coliform of cattle and is excreted in feces. Ingestion of contaminated food and water leads to infection by the Shiga toxin-producing bacteria. Following ingestion, Shiga toxin binds to globotriaosyleramide (Gb3) on endothelial cells, renal mesangial cells, and epithelial cells.27,28 Gb3 is highly expressed in renal endothelial cells, a fact that is thought to explain renal involvement of the disorder.
After binding Gb3, Shiga toxin enters the cell by endocytosis, where it inactivates the ribosome, ultimately leading to cellular apoptosis.28 Shiga toxin also leads to a prothrombotic and pro-inflammatory state that results in early platelet activation and thrombosis.25,28 Microvasculature thrombi develop and result in end organ ischemia, damage, and the resultant kidney injury that is seen in HUS. Thrombi also are thought to contribute to the development of schistocytes and microangiopathic hemolytic anemia.
aHUS results from dysregulation in the alternative complement pathway. In this pathway, C3 is activated in the plasma and deposited on all cellular surfaces in contact with plasma. This includes native host cells. Through a series of steps, the membrane attack complex (MAC) is formed. The MAC causes lysis of the involved cell. This process is regulated by a series of proteins, including CFH, that inactivate C3 and other downstream components and prevent host damage. In aHUS, one or more of these regulator proteins are decreased or absent, leading to progressive activation of the alternative complement pathway and damage to host cells. The persistent complement activation leads to platelet activation and formation of thrombi through several postulated methods.25 Additionally, red blood cells are directly damaged when they come into contact with the complement pathway.
Secondary HUS is less understood and occurs as a result of an ongoing disease process. Cases have been described from various inciting events. The underlying pathophysiology may result from direct endothelial damage, complement activation, autoantibody formation, or complement regulator protein dysregulation.29
Clinical Presentation and Diagnosis
ST-HUS presents in a child following a diarrheal illness. In many of these cases, diarrhea will be bloody as a result of E. coli O157:H7 infection. In fact, 78% of all HUS cases will have a component of bloody diarrhea.23 Diarrhea typically starts three to eight days after consumption of contaminated food or water. On average, ST-HUS is diagnosed six days after the start of diarrhea.28
Patients with HUS may present with signs and symptoms of thrombocytopenia, anemia, or renal failure, including bruising, bleeding, thrombosis, fatigue, pallor, hypertension, oliguria, and anuria. Neurologic causes are evident in 25% of cases.28,30
A high degree of suspicion should be given to patients with these signs or symptoms after hemorrhagic colitis. In aHUS, the absence of diarrhea may make diagnosis more difficult, and reliance on a laboratory workup in the setting of the appropriate clinical context may be necessary.
Diagnosis of HUS is made clinically. A diagnosis of ST-HUS is made when the constellation of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury present following prodromal diarrhea. Diagnosis may be aided by stool or serum testing for Shiga toxin-producing E. coli.
The diagnosis of aHUS is more difficult, as the prodromal diarrhea is not present. The diagnosis of aHUS can be considered in a patient of the appropriate age group with the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. This is confirmed when dysregulation of the complement pathway is demonstrated. The diagnosis of aHUS is unlikely to be made in an ED.
Differential Diagnosis
The clinical manifestations of HUS include anemia, thrombocytopenia, acute kidney injury, hypertension, and neurologic deficits, so the differential diagnosis of the process is long. The differential includes other TMAs and coagulopathies, including TTP, DIC, immune thrombocytopenia (ITP), and heparin-induced thrombocytopenia (HIT). Malignancy also can produce similar presentations.
Infectious agents, such as hepatitis, HIV, Salmonella, and other infectious colitis, can present similarly with anemia, kidney injury, and thrombocytopenia. Likewise, inflammatory bowel disorder, lupus, and other autoimmune disorders may have similar or overlapping presentations with HUS. The differential diagnosis should be tailored to patient-specific presentation and evaluated appropriately within this context.
Management
The management of HUS initially is supportive in all three subtypes.29 Care should be taken to manage dehydration with IV fluids. Careful monitoring of urine output is essential, as decreased output may be an indicator of renal failure and need for dialysis. Electrolyte abnormalities should be managed medically as indicated. Anemia can be managed through transfusion of packed red blood cells. Generally, platelets are indicated only in the event of clinically significant bleeding or for invasive procedures.
As in other processes, dialysis is indicated for anuria, electrolyte abnormalities refractory to medical management, volume overload, acidosis, or azotemia. Hypertension, hypertensive emergency, and seizures should be managed according to current standards of care. As this is principally a pediatric problem, transferring the patient to a pediatric hospital or hospital with resources for pediatric dialysis may be indicated.
Paradoxically, antibiotic administration of a Shiga-toxin infectious diarrhea does not reduce the risk of developing HUS. One study of 71 children with E. coli O157:H7 diarrhea found that antibiotic therapy increased the risk of developing HUS by 14 times (relative risk [RR] 14.3; 95% confidence interval [CI], 2.9-70.7).31 Two years later, a meta-analysis of 10 studies encompassing 1,121 patients found no association between HUS and antibiotic use (pooled odds ratio [OR], 1.15; 95% CI, 0.79-1.68).32 An updated meta-analysis published in 2016 of 17 studies with a total of 1,896 patients also found no association between antibiotic treatment and the development of HUS (pooled OR, 1.33; 95% CI, 0.89-1.99). However, repeat analysis including only studies with a low risk of bias and those employing an appropriate definition of HUS (five studies with a total of 691 patients) did find an association (OR, 2.24; 95% CI, 1.45-3.46). Thus, the current belief is that antibiotic therapy of Shiga-toxin diarrheal infections in children should be discouraged.33
Prognosis
About 50% of all HUS patients will go on to require dialysis for some length of time during the acute illness.22,23 Despite this high rate of renal failure, the prognosis generally is favorable, with 74% of patients making a full recovery.22 Of those who do not fully recover, the majority of these will remain with only minimal renal dysfunction. Only 3% of patients will go on to have chronic kidney disease stage 2 or higher.22 Diagnosis of HUS is associated with a 4% mortality rate.22,23
Summary
HUS is a rare, often self-limiting, hematologic disorder characterized by anemia, thrombocytopenia, and acute kidney injury. The majority of cases occur in the pediatric population following an acute diarrheal illness. Initial treatment is mainly supportive. Given the high rate of acute renal failure, management in a pediatric hospital may be indicated.
Disseminated Intravascular Coagulation
DIC is a spectrum of abnormal coagulation consisting of both thrombosis and bleeding. It can be rapid and life-threatening or chronic and indolent. In the ED setting, the acute presentation creates the most worry. Typically, patients will present with some form of hemorrhage; however, 5-10% of patients can present as isolated microthrombi.34 Perhaps most importantly, DIC is not a primary disease process, but it is secondary to several other pathologic states.
Epidemiology/Etiology
Up to 1% of hospitalized patients will develop some degree of DIC.35 It is seen evenly among men and women. There are multiple underlying triggers, with sepsis being the most common. About 30-50% of sepsis cases caused by gram-negative or gram-positive organisms will progress to DIC.36 The condition is especially likely to develop in patients who are immunocompromised and asplenic, as they will have more difficulty clearing bacteria from the bloodstream.37
Both solid and hematologic malignancies have been associated with DIC. It appears to be associated most commonly with acute myeloid leukemia, occurring in more than 30% of cases.38 Other commonly seen causes include trauma and burns. DIC also can be associated with several obstetric complications, such as placental abruption and amniotic fluid embolism. More rare causes include vascular abnormalities, such as aortic aneurysms, allergic reactions, and immunologic reactions.
Pathophysiology
The mechanism of DIC has been an evolving area of study. Current research supports an initial hypercoagulable state. The activation and release of tissue factor (TF) is one of the main mediators of hypercoagulation in DIC.39 The release of TF from the vessel wall activates factor VII, which then propagates the common pathway of the coagulation cascade.
The anticoagulation systems that typically inhibit thrombin generation (protein C, antithrombin [AT], and tissue factor pathway inhibitor) all are impaired in DIC.40 The protein C system does not function adequately in DIC, mainly as a result of decreased levels of protein C and protein S. Additionally, there is a downregulation of the endothelial protein C receptor.41
In DIC, AT levels are diminished significantly secondary to decreased synthesis, increased breakdown by neutrophil elastase, and excessive consumption due to continuing thrombin production.42 The continuous, widespread activation of the coagulation cascade eventually leads to exhaustion of coagulation factors and uncontrollable hemorrhage.
Conversely, the forms of DIC associated with acute promyelocytic leukemia and some forms of prostate cancer will present early with significant hemorrhage due to a hyperfibrinolytic state not seen with other causes.43,44
Clinical Features
The hallmark of DIC involves thrombosis and bleeding. Ultimately, the presentation varies depending on the underlying cause. DIC brought on by sepsis, obstetric complications, multisystem trauma, and immunologic reactions is more likely to develop acutely and present with evidence of multi-organ dysfunction.37 Significant bleeding, including oozing at sites of vascular access, is seen in severe cases.34
Alternatively, subacute to chronic DIC typically has few, if any, clinical manifestations and is identified only by the presence of laboratory abnormalities.45 Most commonly, this develops secondary to prolonged hypercoagulability, such as that seen with malignancy.37
Diagnostic Studies
When there are manifestations of active bleeding or thrombosis, a full coagulation profile should be obtained. This includes a CBC, prothrombin time (PT), activated partial thromboplastin time (aPTT), and quantification of the fibrin system. There is no one test that can establish DIC, but diagnosis requires the combination of several laboratory abnormalities.
For simplicity, the International Society of Thrombosis and Hemostasis developed a DIC scoring system (see Table 3) based on the common laboratory abnormalities of DIC.47 In this system, a score greater than 5 is consistent with DIC. Several studies have assessed the applicability of the scoring system, with one prospective trial showing 91% sensitivity and 97% specificity when compared to expert opinion.48
Table 3. DIC Scoring System
|
Presence of condition associated |
No |
0 |
|
Yes |
2 |
|
|
Platelet count |
> 100,000/µL |
0 |
|
< 100,000/µL |
1 |
|
|
< 50,000/µL |
2 |
|
|
Fibrin marker (D-dimer) |
No increase |
0 |
|
Moderate |
1 |
|
|
Strong |
2 |
|
|
Prolonged prothrombin time (PT) |
< 3 seconds |
0 |
|
> 3 seconds and < 6 seconds |
1 |
|
|
> 6 seconds |
2 |
|
|
Fibrinogen |
< 1 g/L |
0 |
|
> 1 g/L |
1 |
|
|
Total score ≥ 5 associated with overt DIC Adatped from Bakhtiari K, Meijers J, de Jonge E, Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med 2004;43:2416-2421. |
||
The CBC is used to assess for anemia and thrombocytopenia. A low platelet count (below 100,000/µL) is seen in the majority of cases of DIC, but it is by no means specific for the diagnosis. About 10-15% of patients will have counts less than 50,000/µL.48 This is an important threshold, as lower levels are associated with a four to five times increased risk for hemorrhage.49
DIC is characterized as a consumptive coagulopathy, so it stands to reason that there will be abnormal coagulation profiles. Prolonged PT and aPTT studies are characteristic of DIC and will begin to develop after about a 50% reduction in clotting factor levels.48 Assessment of fibrin also can help to establish the diagnosis. As stated above, in DIC, the fibrinolytic system is impaired, allowing for significant fibrin deposition. D-dimer, a subtype of fibrin degradation products, commonly is assessed and will be elevated in DIC. Again, this marker is not specific, as it will be elevated in a majority of critically ill patients.
While not commonly employed in the ED, thromboelastography (TEG) is used in many ICUs to assess coagulation. Currently, there is limited evidence on the ability of TEG to diagnose DIC, but as research continues, this testing may become more prominent.
Differential Diagnosis
The clinical manifestations of DIC can be seen with a myriad of coagulopathies, so the differential diagnosis is vast. In the setting of severe bleeding, platelet count needs to be assessed. An accurate medication list can be helpful, as platelet number and function can be impaired by heparin (through the development of heparin-induced thrombocytopenia), adenosine diphosphate receptor antagonists, or glycoprotein IIb/IIIa inhibitors.
In patients who appear infected or who have experienced a recent illness, considerations would include hemolytic diseases such as TTP and HUS as well as human immunodeficiency virus (HIV). Individuals receiving mechanical support, such as renal dialysis or extracorporeal membrane oxygenation, also can present with a similar clinical picture. Hematologic malignancy is another important consideration, but differentiation between this and DIC should be possible based on the appropriate laboratory testing.
Management
Because DIC occurs secondary to other disease processes, the mainstay of management involves treating the underlying disorder. DIC presents with several coagulation abnormalities, but it does not appear that transfusion is the answer in all cases. Platelet transfusion should not be considered unless the patient has active hemorrhage with a platelet count below 20,000-30,000/µL or the patient is scheduled for an invasive procedure and the count is below 50,000/µL.39
The use of anticoagulants also appears to be situational. In cases that are caused by vascular abnormalities, metastatic cancer, or purpura fulminans, heparin administration is thought to help prevent propagation of thrombosis.39 As the underlying cause of DIC may not be totally apparent in the ED, it is probably safe to hold off on anticoagulation.
A recent area of study has involved the repletion of natural anticoagulants, such as protein C. Initial literature looked promising, with evidence of improved mortality in patients with sepsis who received protein C; however, follow-up studies did not confirm this and actually showed increased risk of bleeding.39
Disposition
Disposition for patients with DIC will depend heavily on the underlying cause. When there is evidence of end-organ dysfunction or active bleeding, the patient should be admitted. While consultation from the hematology service may be helpful, the patient should be admitted to the service best equipped to manage the underlying condition. For example, trauma and burn patients should be managed by a trauma/general surgery service, while patients with placental abruptions and amniotic fluid embolism should be managed by an obstetrician. The care of DIC patients who present without evidence of bleeding or end-organ dysfunction could be discussed with the patient’s outpatient providers to determine disposition.
Summary
DIC, a consumptive coagulopathy, is characterized by concurrent hemorrhage and thrombosis. This occurs secondary to excessive activation of the coagulation cascade (due to high levels of TF) and impairment of the natural anticoagulation systems in the body. DIC is not a primary disease process. It occurs secondary to many critical illnesses, including sepsis, multisystem trauma, burns, and placental abruption. Providers should have a high index of suspicion when one of its associated underlying causes is present. The management of DIC is mainly supportive, and should focus on treatment of the underlying cause. Platelet transfusion and anticoagulation should be considered only in special cases.
Immune Thrombocytopenia
Introduction
Previously known as idiopathic thrombocytopenic purpura, immune thrombocytopenia (ITP) is characterized by immune-mediated destruction of platelets, which causes a platelet count < 100,000/µL.50 ITP can be a primary disease process or can occur secondary to other autoimmune or infectious conditions. Diagnosis involves review of hematologic laboratory data as well as examination of a peripheral smear. The biggest risk associated with ITP is development of clinically significant bleeding, such as intracranial hemorrhage (ICH). Management has long involved the use of steroids and splenectomy; however, recent studies have examined the use of novel therapies for treatment of ITP.
Epidemiology
In younger adults, ITP is more likely to affect females, while in the elderly population (older than 65 years of age), there is equal incidence between the sexes.51-53 Overall, the annual incidence of primary ITP is 3.3 per 100,000 adults.54 Primary ITP makes up about 80% of the total cases. Recognition and diagnosis of ITP are important because the condition is associated with an increased risk of mortality, with the level of risk proportional to the severity of disease.54 In adults, 80% of cases of ITP will be chronic, lasting longer than one year.55
Etiology
ITP is a diagnosis of exclusion. As stated earlier, 80% of cases are primary ITP, and do not occur secondary to any other condition. The roughly 20% of remaining cases occur secondary to infection, autoimmune disease, hematologic malignancy, or immune deficiency. The most common infections include HIV, hepatitis C, Epstein-Barr virus, and Helicobacter pylori.55 Systemic lupus erythematous, rheumatoid arthritis, and Sjögren’s syndrome frequently are associated autoimmune causes. Non-Hodgkin lymphoma is the most common associated hematologic malignancy, while common variable immunodeficiency is the common immune deficiency causing secondary ITP.55
Pathophysiology
The pathogenesis of secondary ITP depends on the underlying disorder and varies widely. Primary ITP is an acquired immunologic disorder. The condition arises from a combination of antiplatelet antibodies, impaired platelet production, and T cell-mediated platelet destruction.56-58 The contribution of each varies from case to case.
Antiplatelet antibodies are most likely to target surface guanine nucleotide-binding proteins (G proteins). The exact target of the antibodies may influence the course of disease.54 Some research suggests that the production of autoantibodies is due to T cell dysregulation.59 Additionally, T cells can influence the development of ITP directly through CD8+ T cell-mediated platelet lysis.60,61 Decreased platelet production occurs secondary to abnormalities in megakaryocyte growth and apoptosis. An inadequate rise in serum thrombopoietin (TPO) levels also is common and has served as a target of new therapies for ITP.
New research suggests a role of the Ashwell-Morrell receptor (AMR) in the liver in the pathogenesis of ITP. The AMR recognizes older platelets as those that have lost sialic acid from their surface and clears them from circulation.62
Clinical Features
The hallmark feature of ITP is bleeding; however, the majority of patients will present with asymptomatic thrombocytopenia. Risk factors for bleeding include male sex, lower platelet counts (< 30,000/µL), and increasing patient age.54 Common issues include oral petechiae, gingival hemorrhage (see Figure 1), epistaxis, menorrhagia, or occult GI bleeding. Additionally, skin ecchymosis and petechiae (see Figure 2) are expected.
The most feared complication is ICH. There appears to be about a 1.5-1.8% risk of ICH in adult patients with ITP.63,64 Interestingly, patients with chronic ITP are likely to have chronic fatigue, which occurs independently of their platelet count.65
Figure 1. Gingival Hemorrhage in Patient With ITP
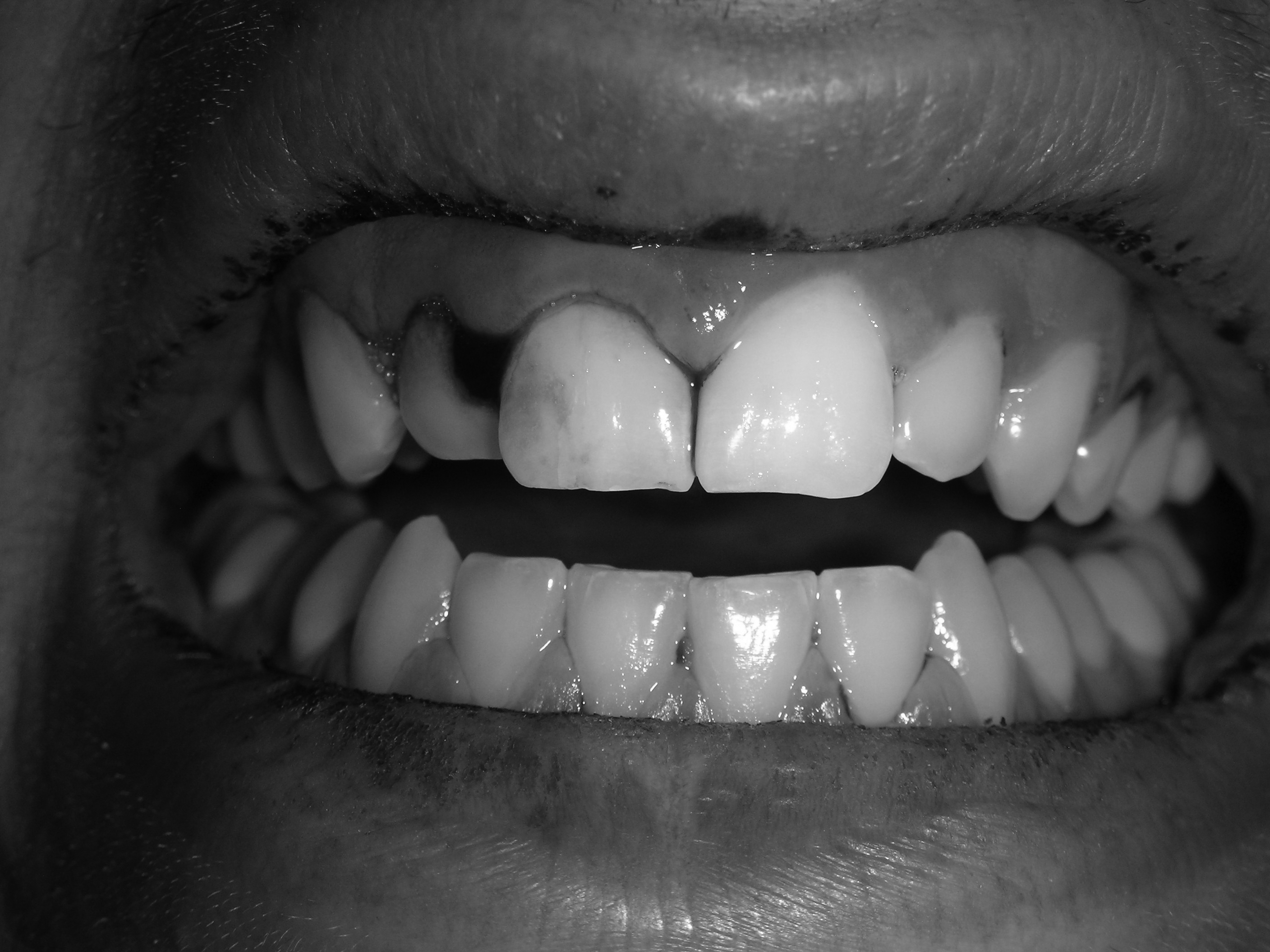
Platelet count was 5,000/µL.
Image courtesy of J. Stephan Stapczynski, MD
Figure 2. Petechiae Purpura in Patient With ITP
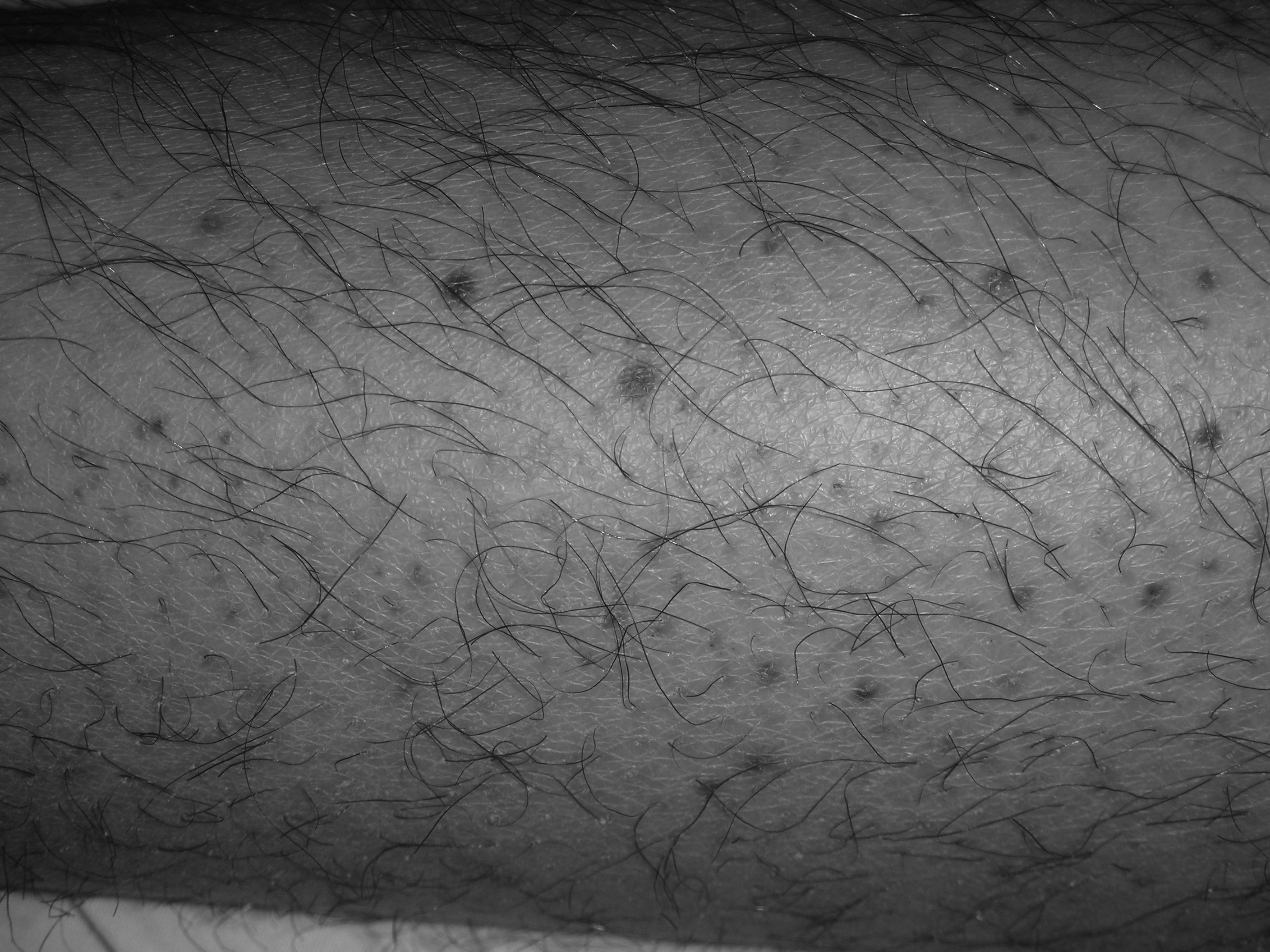
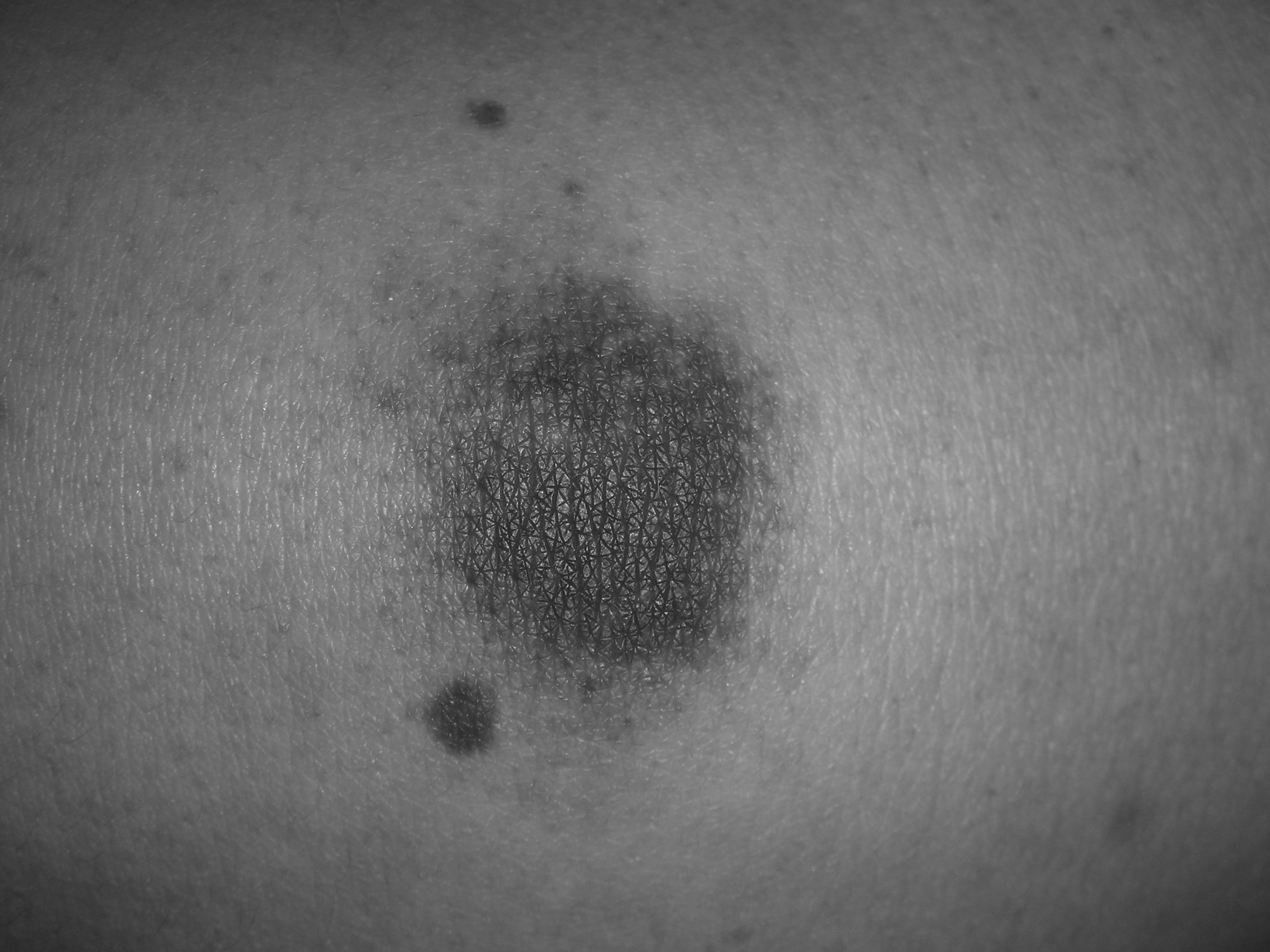
Platelet count was 5,000/µL.
Images courtesy of J. Stephan Stapczynski, MD
Diagnostic Studies
ITP is not a condition that can be diagnosed in the ED. The Internal ITP Working Group (IWG) defines ITP as a platelet count less than 100,000/µL with no identifiable cause.50 Until the appropriate hematologic testing is completed, a final diagnosis cannot be made. For our purposes, the recommendation would be to direct the diagnostic workup based on the clinical findings. Whether there is evidence of mucocutaneous bleeding or concern for ICH, a standard “bleeding” workup should be completed. This would include a CBC, reticulocyte count, peripheral blood smear, coagulation studies (PT, aPTT, INR), and markers of coagulopathy (fibrinogen, D-dimer, etc.). If there is concern for ICH because of changes in mental status or abnormalities in neurological examination, appropriate imaging studies should be obtained (CT scan of the brain).
When a diagnosis of ITP is being considered, the IWG recommends obtaining several additional tests, such as HIV, H. pylori, and hepatitis C antigen testing. These tests likely can be deferred to the admitting team/hematology service.
Differential Diagnosis
ITP is a diagnosis of exclusion, and the differential is very broad. Thrombocytopenia is a commonly encountered abnormality. When patients initially present with isolated thrombocytopenia, it is important to obtain a thorough history and physical examination, as this information may offer clues to the potential cause. Several medications can be responsible, including heparin, digoxin, sulfonamides, aspirin, and phenytoin.66 A history of recent viral infection is a common trigger. A dilutional thrombocytopenia also could be induced in the ED as a result of rapid administration of IV fluids and blood products.
Management
ITP is a disorder of platelet destruction, so, in most cases, new platelets are being produced. These new platelets are more active than older platelets, partially explaining why patients with ITP do not bleed despite often very low platelet counts. Treatment strategies typically are not indicated until the platelet count drops below 20,000 to 30,000/µL or there are manifestations of active bleeding.55 The standard management of ITP involves daily prednisone for two to four weeks, which will induce a transient response in about 80% of patients.54,67 If bleeding is severe, intravenous immunoglobulin (IVIG) can be considered; however, this should not be initiated without consultation with a hematologist.
Splenectomy is the classic treatment in chronic ITP and will induce sustained remission in 60-70% of cases.68 It is no longer commonly employed because of the significant risks associated with asplenia. These include a 30 times increased risk of thrombosis as well as a five to 30 times increased risk of bacterial infection/sepsis in the first 90 days following surgery.54 Replacing splenectomy as second-line therapy are anti-B cell agents and, more recently, the TPO receptor agonists.
Anti-B cell agents, such as rituximab, have resulted in response rates of 40% and 30% at one and two years, respectively.69 They function by inhibiting the plasma cell production of antiplatelet antibodies. TPO receptor agonists (romiplostim and eltrombopag) increase platelet production by megakaryocytes.70 Their initial response rate has been reported as 70-80%.55
While diagnosing ITP in the ED is unlikely, emergency medicine providers may be faced with patients who experience chronic ITP. Taking a good history in these patients is important. If they are post-splenectomy or are taking an immunologic agent, this should raise the suspicion for occult or opportunistic infections.
Disposition
Disposition depends on the clinical scenario. Patients presenting with clinically significant thrombocytopenia need to be admitted for full diagnostic testing because of the significant bleeding risk. Patients with chronic thrombocytopenia with recurrent symptoms who are otherwise well-appearing may be suitable for outpatient management after a conversation with the patient’s hematologist.
Summary
Immune thrombocytopenia is an unexplained platelet count of less than 100,000/µL. The majority of cases are primary and will go on to be a chronic condition. A thorough history and physical examination, as well as appropriate diagnostic testing, will allow for the identification of other likely causes of thrombocytopenia. Acute ITP typically does not require treatment, but when indicated, steroids with or without IVIG are first line. In cases of chronic ITP, splenectomy is being replaced by newer immunologic therapies with good clinical response rates. Ultimately, patients presenting with acute thrombocytopenia require admission to the hospital for diagnostic evaluation and monitoring.
REFERENCES
- Scully M. Trends in the diagnosis and management of TTP: European perspective. Transfus Apher Sci 2014;51:11-14.
- Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010;115:1500-1511; quiz 1662.
- Sadler JE. What’s new in the diagnosis and pathophysiology of thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2015;2015:631-636.
- Mariotte E, Azoulay E, Galicier L, et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): A cross-sectional analysis of the french national registry for thrombotic microangiopathy. Lancet Haematol 2016;3:e237-e245.
- Reese JA, Muthurajah DS, Kremer Hovinga JA, et al. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: Comparison of incidence, demographic and clinical features. Pediatr Blood Cancer 2013;60:1676-1682.
- Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836-2846.
- Terrell DR, Vesely SK, Kremer Hovinga JA, et al. Different disparities of gender and race among the thrombotic thrombocytopenic purpura and hemolytic-uremic syndromes. Am J Hematol 2010;85:844-847.
- Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med 1998;339:1578-1584.
- Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998;339:1585-1594.
- Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017;130:1181-1188.
- George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010;116:4060-4069.
- Adamski J. Thrombotic microangiopathy and indications for therapeutic plasma exchange. Hematology Am Soc Hematol Educ Program 2014;2014:444-449.
- Schwartz J, Padmanabhan A, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical practice — evidence-based approach from the Writing Committee of the American Society for Apheresis: The seventh special issue. J Clin Apher 2016;31:149-162.
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014;371:1847-1848.
- Gottschall JL, Pisciotta AV, Darin J, et al. Thrombotic thrombocytopenic purpura: Experience with whole blood exchange transfusion. Semin Thromb Hemost 1981;7:25-32.
- Harkness DR, Byrnes JJ, Lian EC, et al. Hazard of platelet transfusion in thrombotic thrombocytopenic purpura. JAMA 1981;246:1931-1933.
- Swisher KK, Terrell DR, Vesely SK, et al. Clinical outcomes after platelet transfusions in patients with thrombotic thrombocytopenic purpura. Transfusion 2009;49:873-887.
- Gasser C, Gautier E, Steck A, et al. [Hemolytic-uremic syndrome: Bilateral necrosis of the renal cortex in acute acquired hemolytic anemia]. Schweiz Med Wochenschr 1955;85:905-909. German.
- Javett SN, Senior B. Syndrome of hemolysis, thrombopenia and nephropathy in infancy. Pediatrics 1962;29:209-223.
- Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis 2009;49:1480-1485.
- Flynn JT. Causes, management approaches, and outcome of acute renal failure in children. Curr Opin Pediatr 1998;10:184-189.
- Ardissino G, Salardi S, Colombo E, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr 2016;175:465-473.
- Ong KL, Apostal M, Comstock N, et al. Strategies for surveillance of pediatric hemolytic uremic syndrome: Foodborne Diseases Active Surveillance Network (FoodNet), 2000-2007. Clin Infect Dis 2012;54(Suppl 5):S424-S431.
- Banatvala N, Griffin PM, Greene KD, et al. The United States National Prospective Hemolytic Uremic Syndrome Study: Microbiologic, serologic, clinical, and epidemiologic findings. J Infect Dis 2001;183:1063-1070.
- Jokiranta TS. HUS and atypical HUS. Blood 2017;129:2847-2856.
- Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: State of the art. Mol Immunol 2015;67:31-42.
- Amaral MM, Sacerdoti F, Jancic C, et al. Action of shiga toxin type-2 and subtilase cytotoxin on human microvascular endothelial cells. PLoS One 2013;8:e70431.
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005;16:1035-1050.
- Salvadori M, Bertoni E. Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations. World J Nephrol 2013;2:56-76.
- Gerber A, Karch H, Allerberger F, et al. Clinical course and the role of shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997-2000, in Germany and Austria: A prospective study. J Infect Dis 2002;186:493-500.
- Wong CS, Jelacic S, Habeeb RL, et al. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med 2000;342:1930-1936.
- Safdar N, Said A, Gangnon RE, Maki DG. Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: A meta-analysis. JAMA 2002;288:996-1001.
- Freedman SB, Xie J, Neufeld MS, et al. Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome: A meta-analysis. Clin Infect Dis 2016;62:1251-1258.
- Hunt BJ. Bleeding and coagulopathies in critical care. N Engl J Med 2014;370:847-859.
- Gultawatvichai P, Rathore B. Disseminated intravascular coagulation. In Ferri F, ed. Ferri’s Clinical Advisor 2018: 5 Books in 1. Philadelphia, PA: Elsevier; 2018:398-399.
- Keller TT, Mairuhu AT, de Kruif MD, et al. Infections and endothelial cells. Cardiovasc Res 2003;60:40-48.
- Levi M, van der Poll T. Disseminated intravascular coagulation: A review for the internist. Intern Emerg Med 2013;8:23-32.
- Barbui T, Falanga A. Disseminated intravascular coagulation in acute leukemia. Semin Thromb Hemost 2001;27:593-604.
- Levi M, van der Poll T. A short contemporary history of disseminated intravascular coagulation. Semin Thromb Hemost 2014;40:874-880.
- Levi M, van der Poll T. The role of natural anticoagulants in the pathogenesis and management of systemic activation of coagulation and inflammation in critically ill patients. Semin Thromb Hemost 2008;34:459-468.
- Taylor FB Jr, Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood 2000;95:1680-1686.
- Levi M, van der Poll T, Büller HR. Bidirectional relation between inflammation and coagulation. Circulation 2004;109:2698-2704.
- Avvisati G, ten Cate JW, Sturk A, et al. Acquired alpha-2-antiplasmin deficiency in acute promyelocytic leukaemia. Br J Haematol 1988;70:43-48.
- Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated with acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2-9.
- Seligsohn U. Disseminated intravascular coagulation. In: Handin RI, Lux SE, Stossel TP, eds. Blood: Principles and Practice of Hematology. Philadelphia, PA: JB Lippincott; 2000.
- Toh CH, Hoots WK, SSC on Disseminated Intravascular Coagulation of the ISTH. The scoring system of the Scientific and Standardisation Committee on Disseminated Intravascular Coagulation of the International Society of Thrombosis and Haemostasis: A 5-year overview. J Thromb Haemost 2007;5:604-606.
- Bakhtiari K, Meijers J, de Jonge E, Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med 2004;43:2416-2421.
- Levi M. Diagnosis and treatment of disseminated intravascular coagulation. Int J Lab Hematol 2014;36:228-236.
- Vanderschueren S, De Weerdt A, Malbrain M, et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med 2000;28:1871-1876.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009;113:2386-2393.
- Fogarty PF. Chronic immune thrombocytopenia in adults: Epidemiology and clinical presentation. Hematol Oncol Clin North Am 2009;23:1213-1221.
- Schoonen WM, Kucera G, Coalson J, et al. Epidemiology of immune thrombocytopenic purpura in the General Practice Research Database. Br J Haematol 2009;145:235-244.
- Moulis G, Palmaro A, Montastruc JL, et al. Epidemiology of incident immune thrombocytopenia: A nationwide population-based study in France. Blood 2014;124:3308-3315.
- Lampert MP, Gemsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood 2017;129:2829-2835.
- Audia S, Mahevas M, Samson M, et al. Pathogenesis of immune thrombocytopenia. Autoimmun Rev 2017;16:620-632.
- Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann N Y Acad Sci 1965;124:499-542.
- Khodadi E, Asnafi AA, Shahrabi S, et al. Bone marrow niche in immune thrombocytopenia: A focus on megakaryopoiesis. Ann Hematol 2016;95:1765-1776.
- Olsson B, Andersson PO, Jernås M, et al. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med 2003;9:1123-1124.
- Nishimoto T, Kuwana M. CD4+CD25+Foxp3+ regulatory T cells in the pathophysiology of immune thrombocytopenia. Semin Hematol 2013;50(suppl 1):S43-S49.
- Qiu J, Liu X, Li X, et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci Rep 2016;6:27445.
- Zhao C, Li X, Zhang F, et al. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica 2008;93:1428-1430.
- Rumjantseva V, Hoffmeister KM. Novel and unexpected clearance mechanisms for cold platelets. Transfus Apheresis Sci 2010;42:63-70.
- Neunert C, Noroozi N, Norman G, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: A systematic review. J Thromb Haemost 2015;13:457-464.
- Kühne T, Imbach P, Bolton-Maggs PH, et al; Intercontinental Childhood ITP Study Group. Newly diagnosed idiopathic thrombocytopenic purpura in childhood: An observational study. Lancet 2001;358:2122-2125.
- Arnold DM, Zeller MP, Smith JW, Nazy I. Diseases of platelet number: Immune thrombocytopenia, neonatal alloimmune thrombocytopenia, and posttransfusion purpura. In: Hoffman R, Benz EJ, Silberstein LE, et al, eds. Hematology: Basic Principles and Practice. Philadelphia, PA: Elsevier; 2018:1944-1954.
- Marx JA, Hockenberger RS, Walls RM, et al. Rosen’s Emergency Medicine: Concepts and Clinical Practice. Philadelphia, PA: Elsevier; 2014.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168-186.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623-2634.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura — results of a prospective multicenter phase 2 study. Blood 2008;112:999-1004.
- Kuter DJ. The biology of thrombopoietin and thrombopoietin receptor agonists. Int J Hematol 2013;98:10-23.
Thrombocytopenia is encountered commonly in the emergency department. In most instances, the emergency physician will not be able to determine the definitive diagnosis, but it is important that the initial evaluation be started in a timely manner and that appropriate specialists be consulted from the emergency department.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.