Epilepsy Management in Primary Care
AUTHOR
Sindhu Richards, MD, Assistant Professor of Neurology, Department of Neurology, University of Utah School of Medicine, Salt Lake City
PEER REVIEWER
Padmaja Kandula, MD, Assistant Professor of Neurology and Neuroscience, Comprehensive Epilepsy Center, Weill Cornell Medical College
EXECUTIVE SUMMARY
- Epilepsy affects about 50 million people worldwide, with 5 million people newly diagnosed every year. There is a bimodal distribution, with peaks in the infantile age group and in those older than 60 years of age.
- In adults, the most common cause of epilepsy is a prior stroke. Other frequent causes are brain tumors, infectious etiologies, and chronic alcohol abuse.
- In 2017, the International League Against Epilepsy developed a new classification divided principally between focal and generalized seizures.
- The workup for the first seizure usually includes an electroencephalogram (EEG) and magnetic resonance imaging (MRI).
- Psychogenic nonepileptic spells are more common in females, often with a history of physical/sexual abuse, traumatic brain injury, or medical comorbidities causing distress.
- Drug-resistant epilepsy is defined as the failure of an adequate trial of two tolerated and appropriately chosen anti-epileptic drugs and is an indication for referral to an epilepsy specialist.
- Patients with epilepsy should follow necessary precautions, with most states requiring a three- to six-month seizure-free period before a patient can drive again.
Introduction
Epilepsy affects about 50 million people worldwide and is responsible for up to 0.5% of the global burden of disease. About 7.6 per 1,000 persons will have epilepsy during their lifetime, and there usually is a bimodal distribution, with peaks in the infantile age group and those older than 60 years of age. There are more than 5 million people diagnosed with epilepsy every year and that number is expected to continue to rise.1
Seizures can result from the shift of the normal balance of excitation and inhibition within the central nervous system that may produce a wide variety of clinical symptoms. Epilepsy is a neurological disease defined as having an increased likelihood of recurrent unprovoked seizures.
Etiology
The leading causes of epilepsy vary depending on the age group. In children, febrile seizures are the most common cause of seizures, but these are considered to be provoked seizures and do not fulfill the definition of epilepsy. Generalized idiopathic epilepsy is the most common type of epilepsy in the pediatric population and usually is caused by a genetic etiology, although many of the genetic alterations remain unknown.2 Other causes in the pediatric population may be the result of congenital anomalies or developmental disorders that cause cortical dysplasia or migrational defects. Other, less common etiologies include vascular, hypoxic, traumatic, infectious, neoplastic, or metabolic pathologies.3 Several important epilepsy syndromes to be aware of are included in Table 1.
Table 1. Pediatric Epilepsy Syndromes |
|
|
Age of Onset |
Epilepsy Syndrome |
|
Neonatal |
Benign familial neonatal epilepsy Early myoclonic encephalopathy Ohtahara syndrome |
|
Infancy |
West syndrome Myoclonic epilepsy of infancy Dravet syndrome Benign infantile epilepsy |
|
Childhood |
Genetic febrile seizures plus Lennox-Gastaut syndrome Benign focal epilepsy of childhood Epileptic encephalopathy with continuous spikes and waves during sleep Autosomal-dominant nocturnal frontal lobe epilepsy Landau-Kleffner syndrome Childhood absence epilepsy |
|
Adolescence |
Juvenile absence epilepsy Juvenile myoclonic epilepsy |
In adults, the most common cause of epilepsy is a prior stroke. These strokes are most likely to be caused by ischemic infarction; however, intracranial hemorrhages also can cause epilepsy. The highest risk for seizure is within the first year after a stroke, but the increased risk of seizure continues for several years afterward.4 Another frequent cause of seizures in adults is brain tumors. The glioneuronal tumors, which include gangliogliomas and dysembryoplastic neuroepithelial tumors (DNETs), have the highest incidence of associated seizures. Glial tumors, including glioblastoma multiforme (GBM) and anaplastic astrocytomas, also have a high association with epilepsy. Seizures also can occur with extra-axial tumors, including meningiomas, particularly when there is associated vasogenic edema. There is a lower incidence of epilepsy with metastatic brain tumors compared with the primary brain tumors.5 Infectious causes, including meningitis, encephalitis, or neurocysticercosis, are more common in developing countries. Chronic alcohol abuse is a common cause in adults from 30 to 60 years of age.6 Other causes include idiopathic generalized epilepsy presenting in adulthood, traumatic brain injury, or hypoxic brain injury. Often, as is also the case in children, the cause remains unknown. See Table 2 for a list of the common causes of epilepsy in adults.
Table 2. Common Causes of Epilepsy in Adults |
|
Diagnosis
A seizure can be defined as a paroxysmal alteration of neurological function caused by excessive hypersynchronous neuronal discharges in the brain.7 The clinical symptoms seen during a seizure can vary based on the location of the abnormal electrical discharges, but an individual patient usually has similar symptoms during each seizure. First, it is important to distinguish if the paroxysmal event described by the patient is epileptic or nonepileptic. This entails obtaining a good history from the patient and witnesses about the duration, clinical features of the actual events, and symptoms the patient has immediately before and after the event. The history itself can lead to a diagnosis, but in most cases, other testing will be needed to clarify or confirm the diagnosis.
Certain clinical symptoms can help determine if the episode was a seizure or not. If a patient has an aura, they are usually brief in epilepsy and lasts for a few seconds. Usually seizures last for a total of a few minutes. If the patient consistently describes episodes that are prolonged with retained awareness, then suspicion should be raised about another possible diagnosis. Certain features may point toward a seizure for a generalized tonic-clonic event. These include a loud cry at the beginning, eyes remaining open, foaming or blood at the mouth, lateral tongue biting, and urinary or bowel incontinence. The patients usually will have a postictal period when they remain confused for a few minutes after the seizure. Seizures also tend to be stereotypical, with similar events happening each time. Temporal lobe epilepsy is the most common and affects about 60% of patients who are diagnosed with epilepsy.8 The most common auras in temporal lobe epilepsy are abdominal auras where a patient usually describes a rising epigastric sensation. Psychic auras, including a feeling of fear or déjà vu, and olfactory auras, including distinct smells, also are common in temporal lobe epilepsy.9 The patient also may have impaired awareness and automatisms, which include lip smacking or picking at their blanket. These symptoms also point toward a seizure onset in the temporal lobe. Frontal lobe seizures may begin with focal motor clonic movements on one side of the body because of involvement of the primary motor area. Additionally, frontal lobe seizures also may start with sustained unnatural head version when the seizure originates in the frontal eye field area.9 Hypermotor seizures consist of complex movements involving the trunk and proximal limbs, often during sleep.9 This type of seizure also suggests a frontal lobe origin. Seizures from the parietal lobe may begin with a somatosensory aura (numbness or tingling) that is clearly limited to one defined somatotopic region of the body. Occipital seizures usually begin with a visual aura in a particular visual field.9
Classification of Epilepsy
In 2017, the International League Against Epilepsy (ILAE) commissioned a new classification for epilepsy that eliminated a lot of the previously used terminology.10 (See Table 3.) The first and most important classification to make is whether the onset of the seizure began focally or was generalized. Focal seizures begin in one area of the brain and then may have secondary spread. Next, it is important to distinguish if the patient retained awareness at the beginning of the seizure. This can help to lateralize and localize the seizure onset. After this, focal seizures can be classified further by whether the patient had motor or non-motor symptoms at the onset of the seizure. For example, if a patient does not retain awareness at the onset of the seizure and is smacking their lips or picking at their blanket (automatisms), this would be classified as a focal impaired awareness automotor seizure. This would have been termed a complex partial seizure according to the old classification. The term focal to bilateral tonic-clonic is used when the patient has focal symptoms at the onset of a seizure, but there is subsequent propagation of the seizure to the other hemisphere, causing a tonic-clonic seizure. The term bilateral is used to distinguish it from the classification of generalized seizure, which means that the seizure onset began in both hemispheres.
Table 3. Classification of Seizure Types |
|
Focal Onset Seizures Determine if patient is aware or has impaired awareness
|
|
Generalized Onset Seizures
|
|
Unknown Onset Seizures
|
|
Unclassified Seizures |
|
Adapted from: Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:522-530. |
Generalized seizures are classified further into motor or nonmotor symptoms. For example, generalized nonmotor seizures usually are absence seizures. This diagnosis needs to be confirmed with 3 Hz spike and wave epileptiform discharges seen on electroencephalogram (EEG), as there are some focal impaired awareness seizures that may mimic absence seizures. The classification between focal and generalized epilepsy is important to distinguish. This can alter the treatment plan, because some of the medications are indicated only for certain seizure types and may worsen another seizure type.10
Differential Diagnosis
There are several other important causes of transient paroxysmal events, and it is critical that these are differentiated accurately from epileptic events, since the management will differ. (See Table 4.) Syncope is a transient loss of consciousness caused by a period of inadequate cerebral blood flow. The etiology of syncope can be divided into three causes: reflex, cardiac, or orthostatic. Reflex syncope, previously known as neurogenic, is a transient loss of consciousness because of a reflex response that causes vasodilation and/or bradycardia that leads to systemic hypotension and cerebral hypoperfusion.11 The most common cause of reflex syncope is vasovagal syncope. Orthostatic syncope occurs when there is a significant reduction in blood pressure upon standing caused by impaired autonomic reflex or depletion of the intravascular volume. Syncope also can occur secondary to cardiac arrhythmias. Syncope often presents with characteristic prodromal symptoms, including lightheadedness, sweating, palpitation, and “tunnel vision,” which can help establish the diagnosis. Patients also tend to recover more quickly after a syncopal event than with a seizure. The patient also may have jerking movements while unconscious, called convulsive syncope. These are more likely to be irregular jerks, but at times there can be regular jerking that, to a bystander, may appear to be seizure-like activity.12
Table 4. Differential Diagnosis of Paroxysmal Events |
|
Psychogenic nonepileptic spells (PNES) are paroxysmal changes in the responsiveness or movements of a patient that can resemble an epileptic seizure, but no electrophysiological changes seen. The etiology of PNES is thought to be a subconscious psychological reaction to an underlying stressor. Patients with PNES are more commonly female and often have a history of physical/sexual abuse, traumatic brain injury, or medical comorbidities causing distress.13 Events related to PNES usually are longer in duration than seizures, may differ in symptomatology between episodes, and often do not have a postictal period. However, episodes of PNES sometimes are very hard to distinguish from seizures based on the history alone, and the gold standard is to have the patient admitted for video EEG to capture one of the spells. It has been reported that 20-40% of patients referred to epilepsy monitoring units for refractory epilepsy are found to have PNES.13 Patients with epilepsy also can have PNES, so it is important to verify that all of the episodes they describe are captured on EEG before a definitive diagnosis can be made.
Many other pathologies that cause acute neurologic symptoms can be challenging to differentiate from a seizure. Transient ischemic attacks (TIAs) can cause brief neurological symptoms that, at times, can be mistaken for seizures. TIAs more commonly have negative symptoms, such as loss of motor function, speech, or sensation. Seizures more commonly have positive symptoms, such as clonic movements or paresthesias. Migraines can have auras similar to the ones seen in seizures, but they often are followed by a prolonged headache. Transient global amnesia (TGA) is characterized by a sudden onset of anterograde amnesia that often is accompanied by repetitive questioning that can last up to 24 hours.14 These episodes usually last for hours, unlike the amnesia seen in epilepsy. The pathophysiology of TGA is not well understood at this time.
Certain movement disorders can mimic seizures as well. Myoclonus can be either epileptic or nonepileptic. Nonepileptic myoclonus can be seen in patients with metabolic abnormalities or with spinal cord disease. Some sleep disorders can be misdiagnosed as seizures as well. Rapid eye movement (REM) behavior disorders may be confused for frontal lobe seizures.15 These disorders can be diagnosed with a sleep study, with the movements seen only during REM sleep. Narcolepsy involves periods in which a patient may fall asleep suddenly, which may look like an episode of loss of consciousness concerning for a seizure. The diagnosis of narcolepsy is based on four components: excessive daytime sleep attacks, sudden muscle weakness with emotional symptoms (cataplexy), hypnagogic hallucinations, and sleep paralysis. Taking a careful history to elicit these components can help differentiate it from epilepsy. The definitive diagnosis of narcolepsy can be confirmed with a multiple sleep latency test, which looks for sudden-onset REM on EEG.
Workup of First Seizure
Patients who present with symptoms concerning for a seizure usually will require additional diagnostic workup. The risk for recurrence after an initial seizure is about 32% by one year and 46% by five years.16 However, in elderly patients (≥ 65 years of age), the risk of seizure recurrence after the first unprovoked seizure is up to 80%.17 Certain factors increase the risk of recurrent seizures, including an EEG with epileptiform activity, a nocturnal seizure, or any significant abnormal brain imaging finding.16 After presenting with a first-time seizure, the patient should undergo magnetic resonance imaging (MRI) of the brain and EEG for further workup. MRI with contrast is recommended over computed tomography (CT) imaging because of the insufficient anatomical and soft tissue detail provided by CT. Gadolinium contrast is useful to increase the sensitivity for any tumor that may have caused the seizure. Many centers have epilepsy protocol MRIs that examine the temporal lobes more carefully with thinner cuts. This protocol should be ordered if available. It may help detect subtle findings, such as small areas of cortical dysplasia or mesial temporal sclerosis, that may not be apparent on routine MRI protocols.
The ILAE recently released a new definition of epilepsy. It includes the previous criteria, which state that a patient has to have two unprovoked seizures more than 24 hours apart. Normally, the recurrence rate of another seizure after two seizures is higher than 60%.18 Thus, according to a new criterion from the ILAE, a diagnosis of epilepsy can be made after only one seizure if the probability of having a second seizure is higher than 60%.18 The risk may be elevated to greater than 60% if there is epileptiform activity on an EEG or abnormal imaging findings on MRI. If these are seen, then the patient is considered to have epilepsy and should be started on treatment after the first seizure.
EEG Evaluation
The EEG is one of the most important diagnostic tools for epilepsy to evaluate for abnormal epileptiform activity. A routine EEG, which usually lasts 20-40 minutes, should be the first test ordered in the workup of seizures. A normal EEG does not exclude the diagnosis of epilepsy. More than 40% of patients with epilepsy may have more than one normal EEG, but this can drop to 8% after multiple EEGs with appropriate activating procedures.19 One of the most important activation procedures includes attempting to capture sleep during the EEG. This is attempted by having the patient partially sleep-deprive themselves the night before the study. Adult patients often are also asked to hyperventilate during the EEG, and children may be asked to blow on pinwheels to reproduce hyperventilation. These methods have been shown to activate generalized spike and wave discharges (epileptiform activity) or the absence seizures in more than 90% of patients who have generalized absence epilepsy.19 Intermittent photic stimulation also may be used to help activate interictal activity (abnormal EEG waveforms suggesting increased seizure risk) in juvenile myoclonic epilepsy because light sensitivity may trigger seizures. This is performed by placing a flashing light stimulus in front of the patient and running it at varying frequencies.
If the routine EEG is normal and the patient continues to report uncontrolled seizures, then consider repeating the EEG as a more prolonged study. Ambulatory EEGs, during which the patient can be monitored continuously at home for up to three days, may be an option. If the patient continues to have uncontrolled seizures with a normal EEG, then consider a referral to an epilepsy specialist. Video EEG evaluations in the hospital can be performed at epilepsy centers, which provide much more EEG data. The patient’s antiepileptic drugs (AEDs) may be stopped in the hospital to help increase the yield of the EEG findings. Video EEG evaluations also are very helpful to clarify the diagnosis, especially if there is a concern for nonepileptic spells.
Scalp EEG also has its limitations, since 6 cm2 of cortical discharge is needed to produce a recording.20 If the focus is too small or too deep within the brain, the scalp EEG will not pick up these electrical discharges. For example, seizures from the mesial frontal lobe and insula are not readily seen on the surface unless the seizure propagates to the surface of the brain. This has to be taken into consideration if no EEG changes are seen when a patient is having a paroxysmal events. Additional surface electrodes can be added to localize an epileptiform abnormality more precisely.
Antiepileptic Drug Choices
At this time, there are about 30 AEDs available on the market, with new AEDs currently being developed. (See Table 5.) The ultimate goal in the treatment of epilepsy is to make the patient seizure-free while minimizing the side effects of medication. When initiating treatment for epilepsy, it is recommended to start with monotherapy appropriate for the seizure type. On occasion, the seizure subtype may not be clear. In these cases, one of several broad-spectrum AEDs should be started. Because there are numerous AEDs, the older, previously popular AEDs will be discussed first, then some of the newer AEDs that can be initiated in the primary care office.
Table 5. Antiepileptic Drugs Available on the Market |
|
|
Antiepileptic Drug (Brand Name) |
Year |
|
Phenobarbital |
1912 |
|
Phenytoin (Dilantin) |
1938 |
|
Primidone (Mysoline) |
1954 |
|
Ethosuximide (Zorontin) |
1960 |
|
Diazepam (Valium) |
1968 |
|
Carbamazepine (Tegretol) |
1974 |
|
Clonezepam (Klonopin) |
1975 |
|
Valproic acid (Depakene) |
1978 |
|
Clorazepate (Tranxene) |
1981 |
|
Felbamate (Felbatrol) |
1993 |
|
Gabapentin (Neurontin) |
1993 |
|
Lamotrigine (Lamictal) |
1994 |
|
Fosphenytoin (Cerebyx) |
1996 |
|
Topiramate (Topamax) |
1996 |
|
Tiagabine (Gabatril) |
1997 |
|
Levetiracitam (Keppra) |
1999 |
|
Zonisamide (Zonegram) |
2000 |
|
Oxcarbazepine (Trileptal) |
2000 |
|
Pregabalin (Lyrica) |
2005 |
|
Lacosamide (Vimpat) |
2007 |
|
Rufinamide (Banzel) |
2008 |
|
Vigabatrin (Sabril) |
2009 |
|
Perampanel (Fycompa) |
2011 |
|
Clobazam (Onfi) |
2011 |
|
Eslicarbamazepine (Aptiom) |
2013 |
|
Brivaracetam (Briviact) |
2016 |
|
Cannabidiol (Epidiolex) |
2018 |
|
*XR release versions not included FDA: Food and Drug Administration |
|
Phenobarbital is the oldest AED available on the market. Because of its interactions with other medications and its side effect profile, it is recommended only as first-line therapy for neonatal seizures in the United States. However, because of its efficacy and low cost, the World Health Organization (WHO) recommends it as first-line treatment for focal and generalized tonic-clonic seizures in developing countries.21 Its mechanism of action is that it functions as a gamma-aminobutyric acid (GABA) inhibitory receptor enhancer. When managing a patient’s other medications, it is important to keep in mind that phenobarbital is a strong hepatic enzyme inducer, and the doses of other medications may need to be adjusted accordingly. The half-life of the medication is close to 100 hours, so the dosage needs to be only once daily. Phenobarbital has a narrow therapeutic index, so drug levels must be monitored closely. Barbiturate overdose can cause severe central nervous system depression that can be life-threatening. Patients also may experience abuse or withdrawal with long-term use of the medication. This drug should no longer be initiated as first- or second-line AED therapy in the outpatient setting, as there are several other, safer AEDs.
Phenytoin is another AED that was more popular in the past but is not used as often because of its pharmacokinetics and adverse effects. It is approved for treatment of focal seizures and generalized tonic-clonic seizures. It also is important to note that phenytoin can worsen the seizures in some of the generalized epilepsies, especially myoclonic and absence seizures.10 It blocks the repetitive firing of the voltage-gated sodium channels. At higher doses, the drug follows zero order pharmacokinetics. Because of this, a small increase in the drug dose may increase the serum level significantly and cause toxicity. It is a strong hepatic enzyme inducer, so its interaction with other drugs must be monitored carefully. About 90% of the drug is protein bound; therefore, the drug dose must be adjusted for patients with hypoalbuminemia to avoid toxicity.22 The starting dose usually is 300 mg daily divided into three doses. The therapeutic range is 10 mcg/mL to 20 mcg/mL, and levels should be monitored carefully while adjusting the medication dose. Signs and symptoms that may indicate toxicity include nystagmus, ataxia, dizziness, and dysarthria. Side effects with long-term use include cerebellar atrophy, gingival hyperplasia, osteoporosis, and hirsutism. Phenytoin use should be avoided during pregnancy if possible. It is categorized as a pregnancy category D medication, since it can cause facial cleft and poor neurodevelopment.23
Ethosuximide is approved for treatment of absence epilepsy. It blocks T-type calcium channels, which decreases the burst firing of thalamocortical neurons. It is metabolized by the liver and there is no significant protein binding. The dose usually is started at 500 mg per day and can be increased. The most common side effect is gastrointestinal upset. The half-life of this medicine is 60 hours, but because of its gastric effects, it is given in twice-a-day dosing.
Carbamazepine is approved for focal seizures and generalized tonic-clonic seizures. Like phenytoin, it also can worsen seizures in generalized epilepsies such as myoclonic and absence seizures.10 It inhibits the voltage-dependent sodium channels. Carbamazepine also is a strong hepatic enzyme inducer, like phenobarbital and phenytoin. Carbamazepine also undergoes auto-induction of its own metabolism about two to four weeks after initiation, so the initial dose may need to be increased.24 It has a major metabolite called the 10,11-epoxide, which is responsible for several of the side effects. A baseline sodium level should be checked and then monitored, as carbamazepine can cause hyponatremia. Other side effects include diplopia, ataxia, elevated liver function tests, and osteoporosis. Although a mild rash is a common side effect, it is recommended to exercise caution when starting this medication in the Asian population, who are more likely to have the HLA B*1502 allele, predisposing them to develop Stevens-Johnson syndrome from carbamazepine use.25 This also is a pregnancy category D drug, since it can cause spina bifida and cardiac defects.23
Oxcarbazepine is approved for treatment of focal seizures. It is a structural analog of carbamazepine and has the same mechanism of action. It is metabolized by the liver, but there is no enzyme induction, so it has the advantage of avoiding significant drug interactions. Also, it does not undergo autoinduction and does not form the 10,11-epoxide, avoiding some of the side effects of carbamazepine. The dose is started at 600 mg per day, divided into two doses, and then can be increased. Some of the side effects include gastrointestinal symptoms and headaches. As with carbamazepine, lab work should be monitored for hyponatremia. Hyponatremia is seen more commonly with oxcarbazepine than with carbamazepine. Because of the fewer side effects and fewer drug interactions, oxcarbazepine now is used more commonly now in the outpatient clinic, compared to carbamazepine. There currently is not much data about its risk in pregnancy, since it is a newer agent. The North American Antiepileptic Drug Pregnancy Registry is gathering data about the risk of fetal harm. The clinical decision of whether to continue this drug in pregnancy can be discussed with the patient after weighing the known malformation data.
Valproic acid is approved for broad use as either monotherapy or adjunctive therapy for focal seizures, generalized tonic-clonic, absence, and myoclonic seizures. It works via multiple mechanisms of action, including blockade of sodium channels, blockade of T-type calcium channels, blockade of N-methyl-D-aspartate (NMDA) receptor excitation, and augmentation of GABA release. It is metabolized by the liver and approximately 90% of the medication is protein bound; however, this protein-bound portion decreases as the dosage increases. It also inhibits hepatic enzymes and can increase the levels of other AEDs. The dose usually is started at 250 mg twice per day and can be increased if needed. Valproic acid is a good broad-spectrum drug; however, it has several side effects that limit its use. These include nausea, weight gain, hair loss, tremor, thrombocytopenia, hyperammonemia, pancreatitis, polycystic ovarian syndrome, and, rarely, fatal hepatotoxicity. Valproic acid also is a pregnancy category D drug and can cause severe neural tube defects. In utero exposure has been associated with reduced verbal IQ.26 The teratogenicity is dose-dependent; if a patient has to be maintained on valproic acid, it is recommended to do so at lower doses. The recent trend has been to avoid using valproic acid in women of childbearing age to prevent the potential teratogenicity. There is a reversible parkinsonism and dementia associated with prolonged use of valproic acid in the geriatric population.27
Lamotrigine is approved as adjunctive therapy for focal seizures, generalized tonic-clonic seizures, and seizures associated with Lennox-Gastaut syndrome. It also is approved as monotherapy for focal seizures. Although not Food and Drug Administration (FDA)-approved, it has been shown to be an effective monotherapeutic agent in other generalized epilepsies, including absence and juvenile myoclonic epilepsy.28 Lamotrigine’s mechanism of action includes inhibition of repetitive firing through sodium channel blockade and inhibition of glutamate release. The half-life of lamotrigine can vary significantly based on other medications. When used as monotherapy, the half-life is 24 hours; when used with hepatic enzyme-inducing AEDS, such as phenobarbital, phenytoin, or carbamazepine, the half-life is reduced to 12 hours. When used with enzyme inhibitors, such as valproic acid, the half-life can be extended up to 60 hours. Oral contraceptives and estrogen during pregnancy can increase the lamotrigine metabolism as well. Thus, it is important to monitor the lamotrigine levels in these situations. One possible rare but dangerous side effect is a skin rash that may lead to Stevens-Johnson syndrome. Because of this side effect, lamotrigine is increased very slowly to the therapeutic dose. It usually is started at 25 mg daily and then titrated up by 25 mg to 50 mg every one to two weeks until a goal dosage, usually between 200 mg to 400 mg per day, divided into twice daily dosing, is reached. If a patient is having frequent seizures, this may prohibit the use of lamotrigine, since it can take several months to reach the therapeutic dose. Lamotrigine often is recommended as first-line therapy for women of childbearing age, since it has been well-studied and does not show any major increase in congenital malformations compared to the general population.29
Topiramate is approved as monotherapy or adjunctive therapy for focal and generalized tonic-clonic seizures. It also can be used in myoclonic seizures. Topiramate has several mechanisms of action, which include inactivation of the voltage-sensitive sodium channels, activation of GABA receptors, weak carbonic anhydrase inhibition, and antagonism of the aminomethylphosphonic (AMPA) subtype of the glutamate receptor.30 The drug is excreted primarily through the renal system. At higher doses (greater than 200 mg per day), it induces the cytochrome p450 enzyme and may lower the effectiveness of oral contraceptives.31 The current recommendations are to start at 50 mg per day, divided twice daily, and then increase by 50 mg per week until reaching a goal dose of 200 mg to 400 mg per day. It is contraindicated in patients who have a history of open-angle glaucoma or kidney stones, since it can worsen these conditions. Other side effects include weight loss, cognitive slowing, paresthesias, and oligohydrosis. It is a pregnancy category D drug, since it can cause cleft lip/palate or hypospadias. Thus, the risks vs. benefits of stopping this medication in a pregnant patient should be assessed.23
Zonisamide is FDA-approved as an adjunctive therapy for focal seizures. However, there are several studies that have demonstrated its effectiveness in generalized and myoclonic epilepsies; therefore, it also has been used for these indications.32 It also has been shown to be effective as a monotherapy as well,33 although it is not FDA-approved for this purpose. It has the same mechanism of action as topiramate, with the addition of blocking T-type calcium channels. It primarily is metabolized by the liver, and it has a prolonged half-life of about 60 hours, which allows for once-daily dosing. Unlike topiramate, it has no interactions with other drugs, including oral contraceptives. The most common side effects from zonisamide is dizziness and nausea. Rarely, kidney stones can be seen as well. Although there are limited data, the North American Antiepileptic Drug Pregnancy Registry currently supports that zonisamide monotherapy has a low incidence of malformation. However, caution should be used in continuing this medication for pregnant patients.
Levetiracitam is approved as adjunctive therapy for focal, myoclonic, and generalized tonic-clonic seizures. Although not FDA-approved as monotherapy, clinical trials have shown noninferiority of leveteracitam monotherapy to carbamazepine monotherapy.34 However, levetiracetam has been approved for monotherapy in Europe. Because of this, it sometimes is used clinically as a monotherapeutic agent. It has a unique and incompletely understood mechanism of action, which involves binding to the synaptic vesicle protein 2A (SV2A); the role of SV2A is not entirely known.34 It primarily is metabolized in the kidney, has negligible protein binding, and does not interact with other drugs. The dosing starts at 1,000 mg per day, divided twice daily. However, in clinical practice, it is recommended to start levetiracetam as 250 mg twice a day for a week, then increase to 500 mg twice a day to prevent sedation. Dose adjustments should be made in patients with renal impairment. Behavioral side effects, including irritability and depression, have been reported. These usually occur within the first four weeks of therapy and often improve over time, so it usually is recommended that the patient try to continue the medication for at least one month. Levetiracitam currently is one of the more commonly prescribed AEDs because of its ease of use, broad spectrum, tolerability, and lack of interaction with other drugs.
Lacosamide currently is approved as an adjunctive treatment for focal seizures in patients older than 17 years of age. Lacosamide enhances slow activation of the voltage-gated sodium channels and also binds to collapsin response mediator protein 2 (CRMP2), which is involved in neuronal differentiation and axonal outgrowth.35 Because of the involvement with CRMP2, it also is thought to help with neuropathic pain. Lacosamide is one of the few AEDs that are primarily renally metabolized, and it has no significant interaction with other medications. The dosing starts at 200 mg per day, divided twice daily. However, in clinical practice, it is recommended to start lacosamide as 50 mg twice a day for a week, then increase to 100 mg twice a day to prevent sedation. Lacosamide can cause a small increase in the PR interval on an electrocardiogram that usually has no clinical consequence.36 Clinically, its use is avoided in elderly patients who have a baseline prolonged PR interval. Lacosamide generally is well tolerated, with fewer side effects than some of the other AEDs, and commonly is used by neurologists.
Clobazam is a newer benzodiazepine that is FDA approved as an adjunctive treatment for seizures associated with Lennox-Gastaut syndrome. Studies also have supported its use as adjunctive treatment for both generalized tonic-clonic and focal seizures.37 It is a 1,5 benzodiazepine that enhances GABA activation. The binding site differs from the other benzodiazepines, which results in less sedation. Even with this improvement compared to other benzodiazepines, the main side effect that limits its use is still sedation. It is highly lipophilic and induces hepatic enzymes, so other medications metabolized by the liver should be monitored closely. The dosing starts at 10 mg per day, divided into twice daily dosing, and can be increased slowly.
There has been recent interest in the use of cannabidiol (CBD) in epilepsy. At this time, there is only one FDA-approved CBD oil for clinical use, Epidiolex. It is FDA-approved for patients older than 2 years of age with Lennox-Gastaut or Dravet syndrome.38 The therapeutic dosing usually is around 10 mg/kg per day, and the medication is metabolized by the liver. It can cause up to a threefold increase in the active metabolite of clobazam, desmethylclobazam; therefore, the dosage of clobazam may need to be lowered. Concomitant use with valproic acid can cause significant elevation of transaminases. Patients who want to be considered for Epidiolex treatment likely should be referred to an epilepsy center. Of note, the American Academy of Neurology (AAN) does not support the use of state-sanctioned medical marijuana for neurological disorders at this time, unless there is an FDA approval.39 Medical marijuana is state-dispensed and not FDA-approved. Moreover, because of the psychoactive effect of tetrahydrocannabinol (THC), medical marijuana should be avoided in patients with epilepsy.
Since there are so many AEDs to choose from, sometimes deciding which medication to start can be challenging. If a patient has other comorbidities, choosing an AED that also can help treat their other medical problems can be beneficial. (See Table 6 for examples.) When first considering which AED to start, it is important to choose an agent that will not exacerbate the patient’s seizures and that likely will be effective for the patient’s type of seizure. Although this may seem obvious, if the initial subtype of seizure is unclear, starting with one of several broad-spectrum AEDs often is best. Considering the potential side effects also is important. Even if the medication controls the patient’s seizures, the side effects may be intolerable to the patient and require switching to a new AED. Ease of dosing also is important for patients who have difficulty taking their medications regularly. Extended-release options are available for many AEDs, including levetiracitam, valproic acid, lamotrigine, carbamazepine, and topiramate. In addition, extended-release medications often cause fewer side effects. If an AED fails to control a patient’s seizures, starting a new AED monotherapy with a different mechanism of action likely is the best option. The newer AEDs are generally better tolerated; if a physician continues having difficulty controlling the patient’s seizures, referring the patient to a neurologist is recommended.
Table 6. Antiepileptic Drug Indications and Other Uses |
||
|
AED |
Indications |
Other Uses |
|
Phenobarbital |
Neonatal seizures |
N/A |
|
Phenytoin |
Focal seizures, generalized tonic-clonic seizures |
N/A |
|
Ethosuximide |
Absence seizures |
N/A |
|
Carbamazepine |
Focal seizures, generalized tonic-clonic seizures |
Bipolar disorder, trigeminal neuralgia |
|
Oxcarbazepine |
Focal seizures |
Bipolar disorder, trigeminal neuralgia |
|
Valproic acid |
All seizure types (broad spectrum) |
Bipolar disorder, migraines |
|
Lamotrigine |
All seizure types |
Bipolar disorder, depression |
|
Topiramate |
All seizure types |
Migraines, idiopathic intracranial hypertension |
|
Zonisamide |
All seizure types |
Migraines (less potent than topiramate) |
|
Levetiracitam |
All seizure types |
N/A |
|
Lacosamide |
Focal seizures |
Neuropathic pain |
|
Clobazam |
All seizure types |
Anxiety |
|
AED: antiepileptic drug |
||
Seizure Precautions
Patients with epilepsy should follow several necessary precautions and should be educated regularly about these during office visits. The laws regarding driving vary from state to state, and providers should be knowledgeable about their state’s driving laws. Most states require a three- to six-month seizure-free period before a patient can start driving again; however, some states enforce a seizure-free period of up to one year. There only are five states (California, Delaware, Nevada, New Jersey, Oregon, and Pennsylvania) that have mandatory physician reporting laws to the department of motor vehicles. In most states, patients are expected to report their seizure history to the department of motor vehicles, and their physician will need to fill out a form stating whether the patient may drive. Variation also exists in how states handle patients with seizures that are only nocturnal or that do not affect the patient’s awareness. These restrictions also may vary based on the state. Even if the patient has been seizure-free for the required period of time, the physician should instruct the patient to use caution at times they are more likely to have seizures, including when they are sleep-deprived.
The provider also should discuss water safety precautions with patients with epilepsy. Patients who have epilepsy may be allowed to swim, but they must have one-on-one supervision while swimming. This does not include a lifeguard who is monitoring the whole pool or beach area. They must have an individual watching them the entire time they are swimming to prevent drowning if the patient were to have a seizure. Additionally, it also is recommended that the patient should take showers and not baths. There have been reported cases of drowning as the result of a seizure while bathing.40 Patients who would like to take baths will need one-on-one supervision. It also is important to discuss work safety precautions, such as avoiding operating heavy machinery and working in high places, such as on ladders. The patient should refrain from activities that could result in serious injury to themselves or others if they were to have a seizure.
Intractable Epilepsy
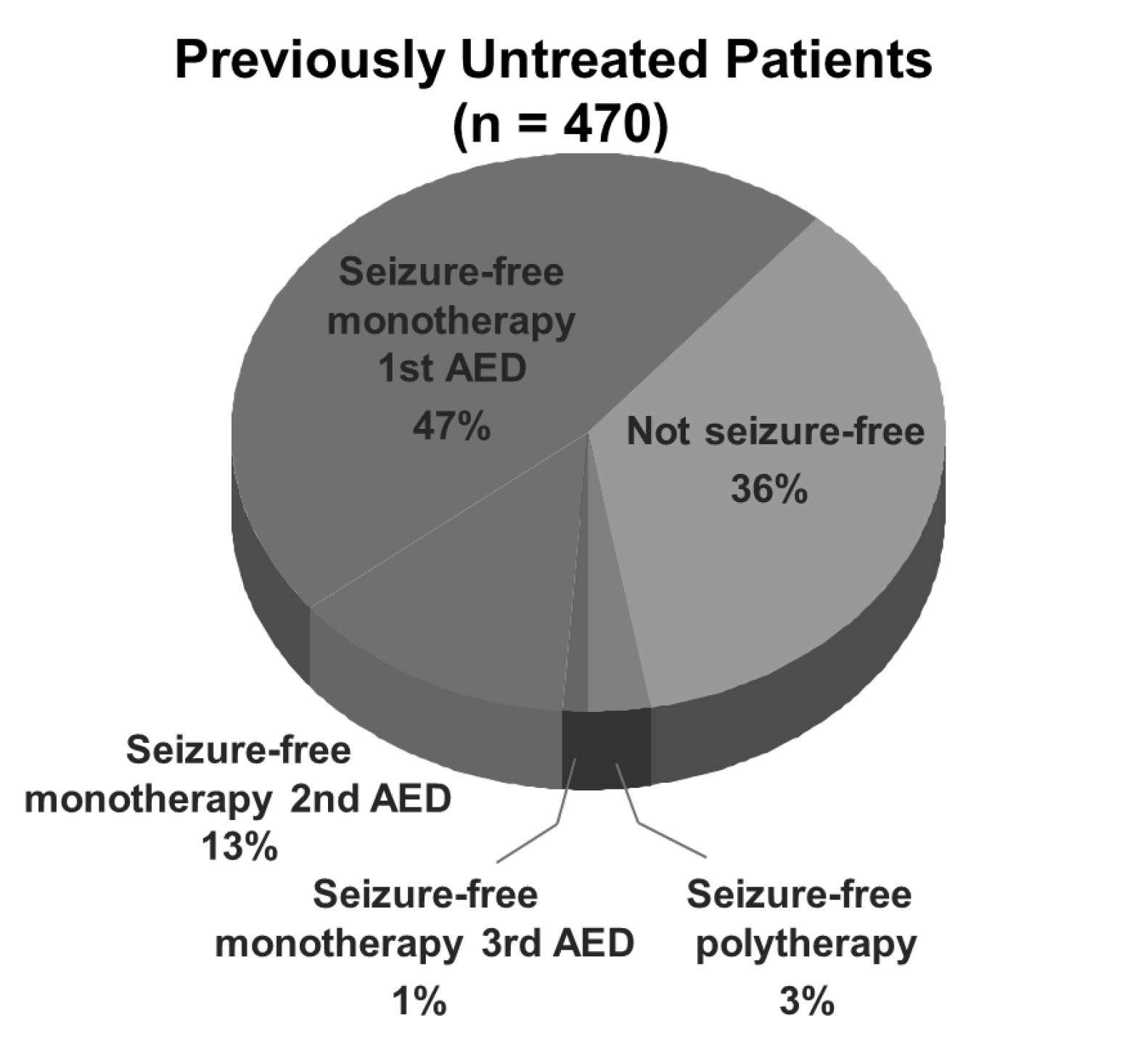
An ILAE task force noted drug-resistant epilepsy may be defined as the failure of adequate trials of two tolerated and appropriately chosen AEDs to achieve sustained seizure freedom.41 In a large study of 470 patients with epilepsy without any prior treatment, 47% of the patients remained seizure-free after initiation monotherapy with their first AED.42 On the other hand, 36% of patients continued to have seizures despite polytherapy with up to three AEDs. (See Figure 1.) These data are similar to other studies, with about half of the epilepsy population responding to the initial AED and about one-third of the epilepsy population developing medically refractory epilepsy. In evaluating these patients, it is important to determine first that the patient is compliant with taking the medication and has no other clear provoking factors for their seizures. After 12 months of unsuccessful treatment by a general neurologist, the patient should be referred to a specialized epilepsy center with an epileptologist.43 In general, a patient who is medically refractory after failing two AEDs also should be referred to an epilepsy center.44 The epilepsy center can provide surgical treatment options, if necessary, and other nonsurgical options, including medications that are not as familiar to general neurologists. Studies have shown that there has been a significant delay in referring a patient for epilepsy surgery. The average duration of epilepsy before surgery is nearly 20 years in almost all trials.45 It is important for primary care providers to identify when patients have medically refractory epilepsy and refer them appropriately to an epilepsy specialist to control seizures as soon as possible.
Figure 1. Percentage of Seizure Freedom on Antiepileptic Drugs |
 |
|
AED: antiepileptic drug |
Surgical Options for Epilepsy
Several surgical options are available at this time for patients with focal epilepsy, several of which were developed in the last several years. Prior to surgery, each patient must undergo an extensive pre-surgical evaluation that starts with a video EEG evaluation. Next, one of several imaging techniques is performed, which may include MRI, positron emission technology/single photon emission computed tomography (PET/SPECT), magnetoencephalogram, and functional MRI, depending on both the epilepsy center and the patient’s clinical situation.46 The patient usually also has neuropsychiatric testing done to help determine language and memory lateralization. This is done to predict if these functions could be affected by surgery. If there is a structural lesion and the EEG data are consistent with the seizures coming from the area, then this lesion usually is resected. However, there often is no clear lesion responsible for the seizures seen on imaging. These cases usually will undergo further workup with invasive EEG evaluation, with depth electrodes placed in the brain area of interest to localize the epileptogenic zone precisely.46
Once the epileptogenic zone is localized, the next step is to decide about the type of surgery. Usually, the epileptic tissue is resected if it is not located in areas of the brain that would cause significant morbidity if resected. For example, resection in epilepsy surgery should be avoided in areas of the brain responsible for language, memory, and motor function. Laser ablation therapy through burr holes in the patient’s skull is a less invasive technique that may be an option, depending on the location and size of the patient’s lesion. This technique is performed using imaging to guide the tip of the ablation catheter to the identified epileptogenic region of the brain, which is then ablated with lasers. Some recent evidence has shown less language and memory problems when using laser ablation compared with open surgery.47
There is a relatively new surgical option called responsive neurostimulation (RNS), which involves a device placed under the scalp, inside the skull. The device has electrodes that record the brain waves and is programmed to detect seizures and deliver an electrical current to stop the seizure. The elecrodes can be placed in areas of eloquent cortex that are not amenable to resective surgery. One of the biggest benefits of RNS is that it provides continuous electrographic data so that the clinician can see the number of seizures. Studies have demonstrated that three out of four patients respond to RNS therapy, achieving > 50% seizure reduction, and one in three patients achieved > 90% seizure reduction.48 Deep brain stimulation (DBS) was approved in 2018 for use in epilepsy. The leads usually are placed in the anterior nucleus of the thalamus, but other areas have been investigated as well, such as the centromedian nucleus and pulvinar. Studies have shown a median seizure reduction of up to about 70% at five years with DBS therapy.49
The surgical therapies mentioned here are only an option for focal epilepsies at this time. For generalized epilepsy, a vagal nerve stimulator (VNS) may be an option. Various studies have been conducted on the efficacy of VNS. In one study, 35% of patients had a seizure reduction of more than 50%, while 20% had a reduction of more than 75%.50 VNS also is approved as an adjunctive therapy for focal seizures. The most common side effects seen with VNS are voice hoarseness, throat discomfort, cough, and paresthesia. There also is a palliative procedure called a corpus callosotomy. This is performed based on the theory that the corpus callosum is required for interhemispheric spread of the epileptic activity. The procedure has been shown to best reduce atonic seizures and often is recommended for patients who have several of these types of seizures.51 It is important to stress to families that the patient likely will not be seizure-free after the VNS procedure.
A randomized, controlled trial demonstrated a 64% chance of seizure freedom with surgery in medically refractory temporal lobe epilepsy, compared with 8% with medical management.52 There also has been evidence that earlier referral for surgery provides a greater chance for seizure freedom in patients with temporal lobe epilepsy.53 Although there is no guarantee that the patient will be seizure-free, it is very important to refer patients to epilepsy centers for surgical evaluation if they are not responding to medications. Most of the patients who undergo surgery are maintained on AEDs, but they usually are able to reduce the number and/or dose of the AEDs, which can improve side effects and their quality of life.
Summary
The diagnosis of epilepsy can be made in the outpatient setting with an appropriate clinical history and diagnostic testing. The first step is to identify if the patient actually is having seizures or if they are having other nonepileptic paroxysmal events. Once the initial seizure is identified, a brain MRI and EEG should be ordered to see if the patient has an increased chance of experiencing recurrent seizures. If this is the case or if the patient has had more than one seizure, then anti-epileptic therapy should be started. The decision of which AED to start depends on the seizure type, side effect profile, and other medical comorbidities. One of the key points for a primary care physician to remember is to recognize patients with medically refractory epilepsy and subsequently refer them in a timely manner to an epilepsy specialist who can offer optimal surgical and nonsurgical options.
REFERENCES
- World Health Organization. Epilepsy: A public health imperative. 2019. Available at: https://www.who.int/mental_health/neurology/epilepsy/report_2019/en/. Accessed Jan. 20, 2020.
- Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet 2009;41:160-162.
- Jan MM. Clinical review of pediatric epilepsy. Neurosciences (Riyadh) 2005;10:255-264.
- Adelöw C, Andersson T, Ahlbom A, Tomson T. Prior hospitalization for stroke, diabetes, myocardial infarction, and subsequent risk of unprovoked seizures. Epilepsia 2011;52:301-307.
- Englot DJ, Chang EF, Vecht CJ. Epilepsy and brain tumors. Handb Clin Neurol 2016;134:267-285.
- Tardy B, Lafond P, Convers P, et al. Adult first generalized seizure: Etiology, biological tests, EEG, CT scan, in an ED. Am J Emerg Med 1995;13:1-5.
- Stafstrom CE, Carmant L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb Perspect Med 2015;5:1-18.
- Blair RDG. Temporal lobe epilepsy semiology. Epilepsy Res Treat 2012;751510.
- Tufenkjian K, Lüders HO. Seizure semiology: Its value and limitations in localizing the epileptogenic zone. J Clin Neurol 2012;8:243-250.
- Bauer J. Seizure-inducing effects of antiepileptic drugs: A review. Acta Neurol Scand 1996;94:367-377.
- Shen W-K, Sheldon RS, Benditt DG, et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2017;70:e39-e110.
- Ozkara C, Metin B, Kucukoglu S. Convulsive syncope: A condition to be differentiated from epilepsy. Epileptic Disord 2009;11:315-319.
- Asadi-Pooya AA, Sperling MR. Epidemiology of psychogenic nonepileptic seizures. Epilepsy Behav 2015;46:60-65.
- Arena JE, Rabinstein AA. Transient global amnesia. Mayo Clin Proc 2015;90:264-272.
- Derry CP, Davey M, Johns M, et al. Distinguishing sleep disorders from seizures: Diagnosing bumps in the night. Arch Neurol 2006;63:705-709.
- Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: Management of an unprovoked first seizure in adults: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Epilepsy Curr 2015;15:144-152.
- Ramsay RE, Rowan AJ, Pryor FM. Special considerations in treating the elderly patient with epilepsy. Neurology 2004;62:S24-29.
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014;55:475-482.
- Panayiotopoulos C. A Clinical Guide to Epileptic Syndromes and Their Treatment. 2nd ed. Springer Healthcare; 2010.
- Ray A, Tao JX, Hawes-Ebersole SM, et al. Localizing value of scalp EEG spikes: A simultaneous scalp and intracranial study. Clin Neurophysiol 2007;118:69-79.
- Ilangaratne NB, Mannakkara NN, Bell GS, et al. Phenobarbital: Missing in action. Bull World Health Organ 2012;90:871-871A.
- Javadi S-S, Mahjub R, Taher A, et al. Correlation between measured and calculated free phenytoin serum concentration in neurointensive care patients with hypoalbuminemia. Clin Pharmacol 2018;10:183-190.
- Hill DS, Wlodarczyk BJ, Palacios AM, Finnell RH. Teratogenic effects of antiepileptic drugs. Expert Rev Neurother 2010;10:943-959.
- Perucca E. Drug interactions with carbamazepine: An ever expanding list? Epilepsy Res 2018;147:119-120.
- Nguyen DV, Chu HC, Nguyen DV, et al. HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in Vietnamese. Asia Pac Allergy 2015;5:68-77.
- Meador KJ. Valproic acid: Reducing the risks of prenatal exposure. Lancet Neurol 2016;15:132-133.
- Armon C, Shin C, Miller P, et al. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology 1996;47:626-635.
- Erksson AS, Nergårdh A, Hoppu K. The efficacy of lamotrigine in children and adolescents with refractory generalized epilepsy: A randomized, double-blind, crossover study. Epilepsia 1998;39:495-501.
- Cunnington M, Tennis P, International Lamotrigine Pregnancy Registry Scientific Advisory Committee. Lamotrigine and the risk of malformations in pregnancy. Neurology 2005;64:955-960.
- Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: Pharmacology, pharmacokinetics, and mechanism of action. Epilepsia 2000;41:S3-9.
- Benedetti MS. Enzyme induction and inhibition by new antiepileptic drugs: A review of human studies. Fundam Clin Pharmacol 2000;14:301-319.
- Kothare SV, Valencia I, Khurana DS, et al. Efficacy and tolerability of zonisamide in juvenile myoclonic epilepsy. Epileptic Disord 2004;6:267-270.
- Kwan S-Y, Chuang Y-C, Huang C-W, et al. Zonisamide: Review of recent clinical evidence for treatment of epilepsy. CNS Neurosci Ther 2015;21:683-691.
- Lyseng-Williamson KA. Levetiracetam: A review of its use in epilepsy. Drugs 2011;71:489-514.
- Perucca E, Yasothan U, Clincke G, Kirkpatrick P. Lacosamide. Nat Rev Drug Discov 2008;7:973-974.
- Rudd GD, Haverkamp W, Mason JW, et al. Lacosamide cardiac safety: Clinical trials in patients with partial-onset seizures. Acta Neurol Scand 2015;132:355-363.
- Humayun MJ, Samanta D, Carson RP. Clobazam. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2019. http://www.ncbi.nlm.nih.gov/books/NBK541043/. Accessed Jan. 22, 2020.
- Sekar K, Pack A. Epidiolex as adjunct therapy for treatment of refractory epilepsy: A comprehensive review with a focus on adverse effects. F1000Res 2019; Feb. 8. [eCollection 2019].
- Koppel BS, Brust JCM, Fife T, et al. Systematic review: Efficacy and safety of medical marijuana in selected neurologic disorders: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2014;82:1556-1563.
- Cihan E, Hesdorffer DC, Brandsoy M, et al. Dead in the water: Epilepsy-related drowning or sudden unexpected death in epilepsy? Epilepsia 2018;59:1966-1972.
- Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51:1069-1077.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000;342:314-319.
- Labiner DM, Bagic AI, Herman ST, et al. Essential services, personnel, and facilities in specialized epilepsy centers—Revised 2010 guidelines. Epilepsia 2010;51:2322-2333.
- Englot DJ, Ouyang D, Garcia PA, et al. Epilepsy surgery trends in the United States, 1990-2008. Neurology 2012;78:1200-1206.
- Haneef Z, Stern J, Dewar S, Engel J. Referral pattern for epilepsy surgery after evidence-based recommendations: A retrospective study. Neurology 2010;75:699-704.
- Lüders H. Textbook of Epilepsy Surgery. 1st ed. Informa Healthcare; 2008.
- Drane DL. MRI-Guided stereotactic laser ablation for epilepsy surgery: Promising preliminary results for cognitive outcome. Epilepsy Res 2018;142:170-175.
- Bergey GK, Morrell MJ, Mizrahi EM, et al. Long-term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology 2015;84:810-817.
- Salanova V, Witt T, Worth R, et al. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology 2015;84:1017-1025.
- Yamamoto T. Vagus nerve stimulation therapy: Indications, programing, and outcomes. Neurol Med Chir (Tokyo) 2015;55:407-415.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008;13:271-278.
- Wiebe S, Blume WT, Girvin JP, et al. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001;345:311-318.
- Engel J, McDermott MP, Wiebe S, et al. Early surgical therapy for drug-resistant temporal lobe epilepsy: A randomized trial. JAMA 2012;307:922-930.
Epilepsy affects about 50 million people worldwide and is responsible for up to 0.5% of the global burden of disease. There are more than 5 million people diagnosed with epilepsy every year and that number is expected to continue to rise.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.