Cardiotoxins: A Systematic Approach to the Evaluation and Management of Patients with Life-Threatening Manifestations of Drug-Induced Cardiotoxicity
Authors: Christopher P. Holstege, MD, FACEP, Director, Division of Medical Toxicology and Blue Ridge Poison Center; Assistant Professor, Department of Emergency Medicine and Pediatrics, University of Virginia, Charlottesville; Alexander B. Baer, MD, Medical Toxicology Fellow, Instructor, Department of Emergency Medicine, University of Virginia, Charlottesville; Mark A. Kirk, MD, Medical Toxicology Fellowship Director, Assistant Professor, Department of Emergency Medicine, University of Virginia, Charlottesville; and William J. Brady, MD, FACEP, Vice Chairman and Associate Professor, Department of Emergency Medicine, University of Virginia, Charlottesville.
Peer Reviewers: Kurt C. Kleinschmidt, MD, FACEP, Assistant Professor, University of Texas Southwestern Medical Center; Associate Director, Department of Emergency Medicine, Parkland Memorial Hospital, Dallas; and Lance Wilson, MD, Department of Emergency Medicine, Case Western Reserve University, MetroHealth Medical Center, Cleveland, OH.
The diagnosis and management of patients with manifestations of drug-induced cardiotoxicity is challenging for even the most experienced emergency physician.
Manifestations of cardiotoxicity are wide and varied, and include bradycardia, QTc prolongation, and life-threatening cardiac dysrhythmias. Although when taken appropriately, in recommended doses, most cardiac agents are safe, susceptible patients, especially those with underlying cardiac abnormalities and/or conduction disturbances, may manifest cardiotoxicity at normal therapeutic doses. When ingested at higher doses in the setting of drug overdose, these agents can produce serious consequences that require precise diagnosis and immediate action. While advanced cardiac life support (ACLS) protocols represent the foundation of management, a number of targeted, pharmacotherapeutic interventions are available that will optimize patient outcomes.1
The following report reviews the pathophysiology and clinical manifestations of cardiotoxins to provide the front-line practitioners with evidence-based protocols for managing patients with life-threatening toxicity.—The Editor
The purpose of this report is to group together agents with similar cardiotoxic effects, review their pharmacologic actions, and discuss the current recommended treatment options of each class. The five main categories reviewed will include: calcium channel blockers, beta-blockers, sodium channel blockers, potassium-efflux blockers, and sodium-potassium adenosine triphosphatase (ATPase) blockers.
Epidemiology
Emergency physicians routinely evaluate and manage poisoned patients. In 2001, more than 2 million human exposure cases were reported to poison centers throughout the United States.2 Of those cases, 22 % (498,524) were treated in a health care facility, with the majority of those cases evaluated in the emergency department (ED). Cardiovascular drugs were listed as the 11th most frequently encountered exposure in adults and the fifth leading cause of poisoning deaths.
Cardiac Physiology
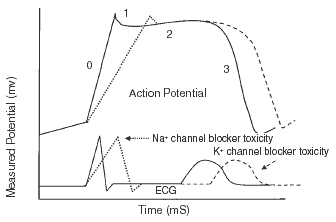
The myocardial cell membrane in its resting state is impermeable to Na+. (See Figure 1.) The Na+-K+ ATPase actively pumps three sodium ions out of cardiac cells while pumping in two potassium ions to maintain a negative electric potential in the myocyte of approximately -90 mV (phase 4). Depolarization of the cardiac cell membrane is due to the rapid opening of Na+ channels and the subsequent massive Na+ influx (phase 0). This Na+ influx causes the rapid upstroke of the cardiac action potential as it is conducted through the ventricles and is directly responsible for the QRS interval of the electrocardiogram (ECG). The peak of the action potential is marked by the closure of Na+ channels and the activation of potassium efflux channels (phase 1). Calcium influx then occurs, allowing for a plateau in the action potential (phase 2) and continued myocardial contraction. The cardiac cycle ends with closure of the calcium channels and activation of potassium efflux channels, causing the potential again to approach -90 mV (phase 3). It is this potassium efflux from the myocardial cell that primarily is responsible for the QT interval of the ECG.
|
General Management of Cardiac Toxins
All patients who present with toxicity or potential toxicity following ingestion of cardiotoxins should be managed aggressively. The patient’s airway should be patent and adequate ventilation assured. If necessary, endotracheal intubation should be performed. Too often, physicians are lulled into a false sense of security when a patient’s oxygen saturation is adequate on high flow oxygen. If the patient has either inadequate ventilation or a poor gag reflex, then the patient may be at risk for subsequent CO2 narcosis with worsening acidosis or aspiration. Because laryngoscopy has been reported to induce a vagal response, the physician may consider administration of atropine in the bradycardic patient prior to intubation. The initial treatment of hypotension consists of intravenous fluids. Close monitoring of the patient’s pulmonary exam should be performed to assure that pulmonary edema does not develop as fluids are infused. The health care providers should place the patient on continuous cardiac monitoring with pulse oximetry and make frequent neurological checks. Placement of a urinary catheter should be considered early in the care of symptomatic patients to monitor urinary output, as this is one of the best indicators of adequate perfusion. In all patients with altered mental status, the patient’s glucose should be checked. These patients should receive a large-bore peripheral intravenous line, and all critically ill patients should have a second line placed either peripherally or centrally. If the patient is a potential candidate for an intravenous pacemaker, a central line should be placed preferentially in the right internal jugular.
Gastrointestinal decontamination should be considered only after initial supportive care has been provided and airway control has been assured. Activated charcoal (1 g/kg) may be administered. Because many cardiotoxins have sustained release preparations, multidose charcoal administration (1 g/kg first dose and then ½ g/kg q 4 hours) should be considered along with whole bowel irrigation (polyethylene glycol with electrolytes at 500 mL/hr for children and 2 L/hr for adults). Syrup of ipecac should not be administered in the ED and is contraindicated after overdose with the agents listed in this paper due to the potential for rapid clinical deterioration. Gastric lavage has not been shown to change outcome after overdose of these agents and can induce an unwanted vagal response.3
Calcium Channel Blocker Toxicity
Definition. There are nine cardiac calcium channel blockers that have been approved for clinical use in the United States. These nine are found within four classes of compounds. (See Table 1.) During the past decade, the number of exposures to these agents has increased dramatically as these drugs have become available on the market. In 2001, calcium channel blockers accounted for 40% of all the deaths due to cardiovascular drugs reported to the American Association of Poison Control Centers (AAPCC).2
|

Pathophysiology. All cardiac calcium channel blockers inhibit the voltage-sensitive L-type calcium channel within the cell membrane.4 This channel resides both in heart and smooth muscle cell membranes. The inhibition of this channel prevents movement of calcium from extracellular sites through the cell membrane to intracellular sites. The inhibition of calcium influx in pacemaker cells and within the conduction system results in slowing of conduction and potential heart blocks (1st-3rd degree), bradycardia, and junctional and ventricular escape rhythms. (See Figure 2.) Decreased intracellular calcium within the myocardial cells results in decreased contractility and decreased cardiac output. Blockade of calcium influx within the vascular smooth muscle cells results in vasodilation. Decreased cardiac output coupled with vasodilation may result in profound hypotension. The dihydropyridine class of drugs tends to have a higher affinity for the peripheral vascular smooth muscle cells and have less effect on the cardiac calcium channels. As a result, the dihydropyridine class of agents more often is associated with hypotension with the possibility of reflex tachycardia. Verapamil and diltiazem, on the other hand, have strong affinity for both cardiac and vascular calcium channels and, subsequently, the combination of hypotension with bradycardia may be seen.
|
The calcium channel blockers also have been associated with profound hyperglycemia refractory to standard doses of insulin.5 Though the exact etiology of this effect is unclear, both the blockade of insulin release and the blockade of peripheral insulin receptors have been suggested as possible mechanisms.6 This hyperglycemic effect has been seen in both verapamil and diltiazem poisoning, but it is not well demonstrated in the dihydroperidine class of calcium channel blockers.
In both animal models and human case series, calcium channel blockers also have been associated with cardiac sodium channel blockade.5,7 This results in a delay of phase 0 of depolarization, subsequent QRS prolongation, and further potential for dysrhythmias (see the "Sodium Channel Blocker Toxicity" section below).
Clinical Features. Hypotension is the most common sign associated with toxicity from a calcium channel blocker. This sign initially may be subtle, with the patient simply complaining of feeling lightheaded or dizzy, especially upon standing. Depending on the agent ingested, reflex tachycardia may not be seen. As levels of calcium channel blocker increase, the patient may develop marked hypotension, various heart blocks, bradydysrhythmias, pulmonary edema, acute mental status changes, syncope, asystole, and death. Hypoperfusion may result in ischemia in patients with pre-existing atherosclerotic vascular disease. Patients may develop myocardial infarction, mesenteric ischemia, seizures, renal failure, strokes, and lactic acidosis.
Diagnostic Studies. Patients suspected of having calcium channel blocker toxicity should have an ECG performed and appropriate laboratory values drawn. Acute toxicity may be associated with hyperglycemia, and a rapid glucose check should be obtained. A chest x-ray should be performed on any patient with suspected pulmonary edema. Serum levels of calcium channel blockers cannot be obtained in a timely manner and are not useful in the initial management of the overdose.
Management. A symptomatic acute calcium channel blocker overdose can be one of the most challenging poisonings encountered by a physician. The initial management of all patients presenting after an acute overdose of a calcium channel blocker or presenting with significant toxicity should be treated as discussed in the above section entitled "General Management of Cardiac Toxins." Calcium channel blockers are prone to development of pulmonary edema, and frequent lung reassessments should be made.8 Specific pharmacological therapy includes the possible use of atropine, calcium, glucagon, insulin, and/or various catecholamines.
Atropine. Atropine may be considered in an attempt to reverse bradycardia at doses of 1-2 mg intravenous (0.02 mg/kg in children, minimum of 0.10 mg and a maximum of 1 mg). In calcium channel blocker intoxications, atropine may be ineffective at reversing bradycardia, and the clinician should not be surprised to see a lack of response to this agent.9
Calcium. In symptomatic calcium channel blocker poisonings, intravenous calcium should be considered early in the care. The exact mechanism is unknown, but a calcium infusion certainly will increase extracellular calcium, thereby increasing the concentration gradient across the cell membrane. This gradient probably drives more calcium intracellular when unblocked calcium channels open. Either calcium gluconate or calcium chloride may be infused through peripheral line access. Recall that 10 mL of 10% calcium gluconate (93 mg Ca2+ or 4.6 mEq Ca2+) provides less bioavailable calcium than 10 mL of 10% calcium chloride (272 mg Ca2+ or 13.6 mEq Ca2+). However, calcium chloride has been reported to cause marked tissue necrosis if it extravasates while being infused through peripheral venous access, so it is best administered through a central venous line. The exact infusion doses of calcium and targeted calcium level have not been well delineated.10 An initial dose of 10 cc of 10% calcium chloride or 1 g (20 mg/kg or up to 1 g in children), or the equivalent dose of calcium gluconate, administered intravenously, is appropriate. This dose may be repeated and titrated to clinical effect. It is a reasonable approach to infuse calcium to increase the ionized calcium 2-5 times the patient’s baseline. However, increasing the patient’s ionized calcium may lead to further sedation, nausea, and vomiting. To assure adequate ventilation and gag reflex, frequent neurological examinations of patients receiving calcium infusions are especially imperative if the patient is not intubated.
Glucagon. Glucagon may be beneficial in the management of the calcium channel blocker overdose.11,12 Glucagon raises intracellular cyclic adenosine monophosphate (cAMP) concentrations that in turn open calcium channels. A starting dose of 2-5 mg (50 mcg/kg in children) of intravenous glucagon (diluted in 10 cc normal saline [NS] and infused over 1-2 minutes) should be considered. This dose can be repeated. If the patient responds to the initial glucagon dose, then a glucagon infusion at 5 mg/hr (50 mcg/kg/hr in children) should be started. Glucagon doses this high may induce emesis that could lead to aspiration in the sedate, non-intubated patient. When mixing glucagon, use NS and avoid using the glucagon package diluent. The package diluent contains 0.2% phenol and hypothetically could result in toxicity at doses this high.
Catecholamines. The role of the catecholamines is unclear. Catecholamine infusions may be considered after the above therapies fail to give adequate response. Both epinephrine and norepinephrine have been used in the management of calcium channel blocker toxicity with mixed success. Care should be exercised, as these agents may result in exacerbation of pulmonary edema, ischemic vascular disease, and renal failure. Dopamine also should be infused with caution due to its poor direct effects on adrenergic receptors compared to epinephrine and norepinephrine. Dobutamine, a direct beta-1 adrenergic agonist, may be of benefit, but adequate studies of its effect in calcium channel blocker toxicity are lacking.
Insulin. High-dose insulin drips have been advocated in calcium channel blocker overdose, especially in the management of both verapamil and diltiazem poisoning. Experimental models suggest a superior effect of this therapy in the canine model compared to other therapies.13 Human case reports also suggest the efficacy of these drips.5,14,15 The optimal dose of insulin is unclear.16 Insulin infusions can be initiated at 0.2 U/kg/hr and titrated upward every hour to euglycemic effect and hemodynamic effect. Supplemental glucose may be necessary to maintain euglycemia. Drips exceeding 100 units per hour have been performed to achieve euglycemia. Serum glucose and potassium levels should be monitored closely during therapy.
Sodium Bicarbonate. Recently, sodium bicarbonate (SB) infusions have been advocated in the management of calcium channel blocker toxicity.5,7 Patients with prolonged QRS (> 100 ms), acidosis, or persistent hypotension despite the above methods should be considered candidates for a trial of SB in doses noted in the section titled "Sodium Channel Blockers."
Other Therapies. Pacemakers, intra-aortic balloon pump, and cardiopulmonary bypass all may be considered in cases not responding to pharmacological therapy. Vasopressin recently has been reported to reverse hypotension resulting from specific overdoses, but there are no human or animal reports of its use in calcium channel blocker toxicity. None of the calcium channel blockers are amendable to hemodialysis or hemoperfusion due to their large size, high protein binding, and/or large volume of distribution.
Disposition. Asymptomatic calcium channel blocker overdose patients with normal ECGs and ingestions of non-sustained release products may be observed in the ED for eight hours. If they remain asymptomatic and the ECG remains unchanged, they may be discharged to a non-monitored setting. Any asymptomatic patient acutely overdosing on a sustained release product should be admitted to a monitored bed for 24 hours of observation. Any symptomatic calcium channel blocker patient should be admitted to a monitored setting until complete resolution of symptoms occurs. Care should be exercised in patients with apparent improvement but who remain symptomatic following calcium channel blocker overdose, as sudden asystole has been reported hours following stabilization and apparent improvement.
Beta-Blocker Toxicity
Definition. Beta-adrenergic receptor antagonists increasingly are used due to their efficacy in the treatment of hypertension, ischemic heart disease, and arrhythmias. There currently are numerous beta-blockers available. (See Table 2.) These agents share the mechanism of competitive beta-adrenergic receptor antagonism. Some of these agents have equal affinity for beta-1 and beta-2 receptors (propranolol), while others are selective and have greater beta-1 than beta-2 receptor blocking activity (metoprolol). Some agents also block other receptors such as alpha-adrenergic receptors (labetalol), cardiac sodium channels (propranolol), and cardiac potassium efflux channels (sotalol).17
|

Pathophysiology. Beta-blockers competitively inhibit the beta-adrenergic receptor. Inhibition of beta-1 receptors results in a decrease in the force and rate of myocardial contraction, a decrease in AV node conduction velocity, and a decrease in renin secretion. Inhibition of beta-2 receptors results in a decrease in glycogenolysis, decrease in gluconeogenesis, and decrease in relaxation of smooth muscles in blood vessels, bronchi, and the gastrointestinal tract.
Clinical Features. In acute overdose, the most pronounced effect of beta-blockers is on the cardiovascular system.12 Bradycardia, heart blocks, and hypotension are the hallmarks of significant beta-blocker toxicity.18 Dyspnea may occur and may be the result of congestive heart failure or bronchospasm. Changes in mental status, seizures, and coma have been reported and typically are associated with coexisting cardiovascular effects and hypoperfusion. Hypoglycemia may occur and should be considered in those patients with altered mental status or seizures, especially young children.
Diagnostic Studies. Patients suspected of having beta-blocker toxicity should have an ECG performed and appropriate laboratory values drawn. Acute toxicity may be associated with hypoglycemia, and a rapid glucose check should be obtained in any patient presenting with acute mental status changes. Serum levels of beta-blocker cannot be obtained in a timely manner and are not of clinical utility. A chest x-ray should be obtained in any patient with suspected pulmonary edema.
Management. The initial management of all patients who present after an acute overdose of a beta-blocker or with significant toxicity should be treated as discussed in the above section on "General Management of Cardiac Toxins." Specific pharmacological therapy may include atropine, glucagon, calcium, insulin, and/or various catecholamines.19
Atropine. Atropine may be considered in an attempt to reverse bradycardia at doses of 1-2 mg intravenous (0.02 mg/kg in children, minimum of 0.10 mg and a maximum of 1 mg). In beta-blocker toxicity, atropine has been shown to have poor effect in reversing bradycardia and increasing blood pressure.
Glucagon. Glucagon infusion should be considered in symptomatic beta-blocker toxic patients.12 Beta-adrenergic stimulation raises intracellular cAMP concentrations that in turn regulate ion channels. When beta-adrenergic receptors are inhibited, intracellular cAMP levels decrease. Glucagon increases intacellular cAMP through non-adrenergic pathways.20 A starting dose of 2-5 mg (50 mcg/kg in children) of intravenous glucagon (diluted in 10 cc NS and infused over 1-2 minutes) should be considered. This dose can be repeated. If the patient responds to the initial glucagon dose, then a glucagon infusion at 5 mg/hr (50 mcg/kg/hr in children) should be started. For further information on glucagon therapy, see the section on "Calcium Channel Blocker Toxicity."
Calcium. Calcium has been shown to have efficacy at reversing the hypotensive effects of beta-blocker toxicity in both animal models and human case reports.12,21 Dosing of calcium should be performed at the doses noted in the calcium channel blocker section.
Catecholamines. Catecholamine infusions may be considered after the above therapies fail to give adequate response. Both epinephrine and norepinephrine have been used in the management of beta-blocker toxicity. Care should be exercised, as these agents may result in exacerbation of pulmonary edema, ischemic vascular disease, and renal failure. Dopamine also should be infused with caution. Recall that dopamine at low concentrations (1-5 mcg/kg/min) affects dopamine receptors in renal, mesenteric, and coronary beds, leading to vasodilation. At somewhat higher concentrations (5-10 mcg/kg/min), dopamine exerts positive ionotropic effects by acting on beta-1 adrenergic receptors. It is not until dopamine is infused at high concentrations (greater than 10 mcg/kg/min), that dopamine affects alpha-1 adrenergic receptors and leads to vasoconstriction and elevated blood pressure. Unlike the direct alpha-1 adrenergic agonist activity of epinephrine and norepinephrine, dopamine does not have significant direct activity and instead induces norepinephrine release at the alpha-1 adrenergic receptor. Dobutamine, a direct beta-1 adrenergic agonist, may be of benefit, but adequate studies of its effect in beta-blocker toxicity are lacking. Isoproterenol is a potent, non-selective beta-adrenergic agonist. The literature has reported mixed results with its use and it also should be considered only after the above therapy has failed.
Insulin. Insulin infusions have been advocated for beta-blocker toxicity based on an animal model.22 The exact mechanism of this therapy is unclear, but it is thought to be secondary to increased myocardial glucose utilization resulting from the high-dose insulin drips. Infusions of insulin at 0.5 U/kg/hr along with glucose infusions have been advocated by some clinical studies where the other above methods have failed. Care should be taken to closely monitor blood glucose levels during these infusions.
Other Therapies. The phosphodiesterase inhibitors amrinone and milrinone increase cAMP concentrations and, therefore, theoretically would be useful in beta-blocker toxicity. Current animal models have demonstrated mixed results with use of these agents in beta-blocker toxicity.23,24 Pacemaker insertion, balloon pump, and bypass all may be considered in cases not responding to pharmacological therapy. Extracorporeal removal has been reported with specific beta-blockers with small volumes of distribution and low protein binding (atenolol, nadolol, and acebutolol), but it is technically difficult if there is coexisting hypotension. Vasopressin recently has been reported to reverse hypotension on specific overdoses, but there are no human or animal reports of its use in beta-blocker toxicity.
Disposition. Asymptomatic beta-blocker overdose patients with normal ECGs and ingestions of a non-sustained release beta-blocker may be observed in the ED for six hours.25 If they remain asymptomatic and the ECG remains unchanged, they may be discharged to a non-monitored setting. Any asymptomatic patient acutely overdosing on a sustained release beta-blocker or on sotalol should be admitted to a monitored bed for 24 hours of observation. Any symptomatic beta-blocker toxic patient should be admitted to a monitored setting until complete resolution of symptoms occurs.
Sodium Channel Blocker Toxicity
Definition. The ability of drugs to block cardiac Na+ channels has been well described in numerous previous literature reports, with Kolecki and Curry publishing an excellent review of these agents in 1997.26 This Na+ channel blockade activity has been described as a membrane stabilizing effect, a local anesthetic effect, or a quinidine-like effect. All the agents listed in Table 3 are similar in that they may induce myocardial Na+ channel blockade and may respond to SB therapy.
|

Pathophysiology. Cardiac voltage-gated sodium channels reside in the cell membrane and open in response to depolarization of the cell. The Na+ channel blockers bind to the transmembrane Na+ channels and decrease the number available for depolarization. This creates a delay of Na+ entry into the cardiac myocyte during phase 0 of depolarization. As a result, the upslope of depolarization is slowed and the QRS complex widens. (See Figure 1.)
Clinical Features. Myocardial Na+ channel blocking drugs comprise a diverse group of pharmaceutical agents. As a result, patients poisoned with these agents will have a variety of clinical presentations. For example, cyclic antidepressants, propoxyphene, and cocaine may result in anticholinergic, opioid, and sympathomimetic syndromes, respectively. In addition, these agents may affect not only the myocardial Na+ channels, but also other myocardial ion channels, such as the calcium influx and potassium efflux channels. This may result in ECG changes and rhythm disturbances not related entirely to the drug’s Na+ channel blocking activity.
Sodium channel blockers result in widening of the QRS complex. (See Figure 3.)27 In some cases, the QRS complexes may take the pattern of recognized bundle-branch blocks. In the most severe cases, the QRS prolongation becomes so profound that it is difficult to distinguish between ventricular and supraventricular rhythms.28
|
Continued prolongation of the QRS may result in a sine wave pattern (see Figure 4) and eventual asystole. Sodium channel blockers also may induce a monomorphic ventricular tachycardia. It has been theorized that the Na+ channel blockers can cause slowed intraventricular conduction, unidirectional block, the development of a re-entrant circuit, and a resulting ventricular tachycardia. (See Figure 5a.) This then can degenerate into ventricular fibrillation. Because many of the Na+ channel blocking agents also are anticholinergic or sympathomimetic agents, bradydysrhythmias are rare. However, the Na+ channel blocking agents can affect cardiac pacemaker cells. Bradycardia may occur due to slowed depolarization of pacemaker cells that depend upon entry of Na+. In Na+ channel blocker poisoning by anticholinergic and sympathomimetic drugs, the combination of a wide QRS complex and bradycardia is an ominous sign and may indicate that the Na+ channel blockade is so profound that a tachycardia cannot be mounted in response to muscarinic antagonism or adrenergic agonism.
|

|

Diagnostic Studies. Patients suspected of having toxicity due to a Na+ channel-blocking agent must have an ECG performed and their interval indices should be scrutinized.27 Appropriate laboratory values should be obtained. Specific drug levels are not helpful in the initial management of these poisonings and poorly correlate with ECG findings.
Management. The initial management of all patients presenting after an acute overdose of Na+ channel-blocking agents or presenting with significant toxicity from these agents should be treated as discussed in the above section on "General Management of Cardiac Toxins." Specific pharmacological therapy may include SB infusion.
Sodium Bicarbonate. The management of Na+ channel-blocking agents consists of administration sodium and/or alkalinizing the patient.29 Infusion of SB either by intermittent bolus or by continuous infusion has been advocated. Hypertonic sodium infusion also has been advocated. The indications for SB infusion include a QRS duration of greater than 100 ms, persistent hypotension despite adequate hydration, and dysrhythmias. An ampule of SB contains 50 mEq of sodium and multiple doses may be necessary to achieve clinical improvement in the patient. Start an infusion by adding three ampules of SB to 1 liter of D5W and infuse at two times maintenance. Consider adding potassium (40 mEq to a liter of D5W) to the SB drip to prevent the development of hypokalemia (due to the excretion of potassium in exchange for hydrogen ions as the kidneys attempt to correct the alkalosis). The infusion then can be adjusted as necessary to decrease QRS duration and improve clinical outcome. The exact mechanism of SB therapy is unknown. SB infusions certainly will increase extracellular sodium, thereby increasing the concentration gradient across the cell membrane and probably driving sodium intracellular when unblocked sodium channels open. The benefit of alkalosis has been well demonstrated, but the mechanism is poorly understood. During infusions of SB, close monitoring of electrolyte, pH, and fluid balance should be performed. (See Figure 5b.)
|
Other Therapy. Hyperventilation has been shown to be effective in reversing sodium channel blocking activity, probably due to the respiratory alkalosis induced. Lidocaine has been suggested in the treatment of ventricular dysrhythmias, though clear evidence is lacking. Class IA and IC antiarhythmics should be avoided due to their ability to block cardiac sodium channels.
Disposition. Any symptomatic patient who has ingested a potential Na+ channel-blocking agent should be admitted and observed in a monitored setting. In the asymptomatic patient who has ingested a Na+ channel-blocking agent, the length of observation will vary. For example, a patient who has ingested amitriptyline should be observed for a minimum of six hours, and if that person remains asymptomatic and develops no ECG changes during that time period, then the patient can be discharged. Other poisonings, such as diphenhydramine, can be discharged if asymptomatic after a four-hour observation period.
Potassium Efflux Blocker Toxicity
Definition. Potassium efflux-blocking agents competitively inhibit cellular potassium efflux during cellular repolarization. (See Figure 1.) This results in a delay in repolarization corresponding with an increase in the QT interval. (See Figure 6.)30 This may place the patient at risk for polymorphic ventricular tachycardia or torsades de pointes.31 Some drugs, such as sotalol, are prescribed specifically for this mechanism.32 Other medications possess this activity as an unwelcome side effect at therapeutic doses. A number of medications, such as cisapride and terfenadine, have been removed from the U.S. market due to reports of torsades de pointes and sudden death in patients taking these drugs.33,34 Other medications rarely have been reported to cause QT prolongation except when taken in massive overdose. A complete listing of the reported agents associated with QT prolongation are noted in Table 4.35 Other etiologies of prolongation of the QT interval include: congenital long QT syndrome, hypokalemia, hypomagnesemia, hypocalcemia, myocardial ischemia, neurological catastrophes, and hypothyroidism.36
|

Pathophysiology. The primary problem with K+ efflux channel-blocking drugs is that they prolong cardiac action potentials, which lead to torsades de pointes.37 Patients at increased risk are those who have familial QT prolongation or electrolye abnormalities, or those who take multiple agents with K+ efflux channel-blocking activity or drugs that inhibit the metabolism of K+ efflux channel-blocking agents, resulting in the build-up to toxic levels.
Clinical Features. Syncope may be associated with drug-induced QT prolongation. Palpitations, dyspnea, and sudden death also may be associated with QT prolongation. After acute overdose, the presenting signs and symptoms may not be related to the cardiac effects, but rather to other mechanisms of those drugs.
Diagnostic Studies. Patients suspected of having toxicity due to a K+ efflux channel-blocker should have an ECG performed and appropriate laboratory values drawn to rule out coexisting electrolyte abnormalities. If thyroid disorder is suspected, thyroid function studies also should be performed.
Management. If a patient has a prolonged QT interval due to a K+ efflux-blocking agent, therapy should focus on immediate withdrawal of the potential cause and correction of any coexisting hypoxia or electrolyte abnormalities. All patients initially should be treated as discussed in the above section titled "General Management of Cardiac Toxins."
Magnesium. Intravenous magnesium sulfate is an effective and benign intervention to suppress occurrence of dysrhythmias associated with QT prolongation, even though Mg++ does not typically result in shortening of the interval itself.36 A starting dose of 2 g in 10 mL D5W can be given intravenously and titrated to effect.
Overdrive Pacing. In patients with intermittent runs of torsades not responsive to magnesium therapy, electrical overdrive pacing should be considered.39 Pacing at rates up to 100-120 bpm often is effective at terminating torsades de pointes.
Cardioversion. In the presence of a nonperfusing rhythm, such as ventricular fibrillation, pulseless ventricular tachycardia, or torsades de pointes, unsynchronized electrical defibrillation should be performed.
Disposition. Patients with newly diagnosed prolongation of their QT interval or torsades de pointes should be admitted to a monitored setting. Symptomatic patients who have ingested these agents also should be admitted to a monitored setting.
Sodium-Potassium ATPase Blocker Toxicity
Definition. Cardiac glycosides are agents that inhibit the sodium-potassium adenosine triphosphate (Na+-K+ ATPase) pump. Digoxin is the most widely encountered cardiac glycoside, but numerous other similar-acting agents also exist. (See Table 5.) Digoxin historically has been administered to treat supraventricular tachydysrhythmias and congestive heart failure, but its use has been decreasing as newer agents have been developed. Ingestion of specific plants and contaminated herbal products also has been associated with cardiac glycoside toxicity.40
|

Pathophysiology. The cardiac glycosides inhibit active transport of Na+ and K+ across cell membranes by inhibiting the Na+-K+ ATPase. This results in an increase in both extracellular K+ and intracellular Na+. An increased intracellular Na+ results in a reduced transmembrane Na+ gradient and subsequent increased activity of the Na+-Ca+ exchanger. Intracellular calcium rises, which augments myofibril activity in cardiac myocytes and causes a positive inotropic action. The cardiac glycosides also increase vagal tone, leading to a direct AV depression and conduction disturbances.
Clinical Features. Cardiac glycoside toxicity may result in a wide array of dysrhythmias.41 Excitant activity (atrial, junctional, and ventricular premature beats and tachydysrhythmias), suppressant activity (i.e., sinus bradycardia, bundle-branch blocks, and first-, second-, and third-degree blocks), and combination of excitant and suppressant activity (atrial tachycardia with atrioventricular block and second-degree block with junctional premature beats) all may be seen. (See Figure 7.) The most common toxicity-related dysrhythmia is frequent premature ventricular beats. Bidirectional ventricular tachycardia is stated to be specific for digitalis toxicity but rarely is seen.
|
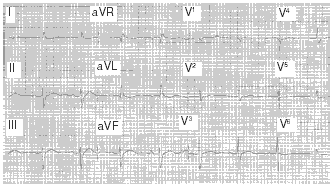
In addition to the cardiac manifestations, non-cardiac signs and symptoms may occur in cardiac glycoside toxicity and vary widely depending on whether the toxicity is acute or chronic. Acute toxicity may have few initial signs and symptoms prior to cardiac instability. Chronic toxicity, on the other hand, may have multiple signs and symptoms, including anorexia, nausea, vomiting, headache, fatigue, depression, dizziness, confusion, memory loss, delirium, hallucinations, and visual disturbances such as seeing yellow halos around objects (xanthopsia). Not uncommonly, chronic digoxin intoxication may be misdiagnosed as a more common illness, such as influenza or gastroenteritis.
Diagnostic Studies. Patients suspected of having cardiac glycoside toxicity should have an ECG performed and electrolytes obtained. Acute toxicity most closely correlates with hyperkalemia, as the Na+-K+-ATPase is inhibited and extracellular K+ rises. In chronic toxicity, hyperkalemia may not be seen. This is due to the slow rise in K+, allowing the kidneys to correct the imbalance. Many of those patients on chronic digoxin therapy also are taking diuretics that further promote potassium excretion. Chronic toxicity associated with renal failure may result in hyperkalemia due to the kidneys’ inability to compensate.
A serum digoxin level should be obtained; therapeutic levels are 0.5-2.0 ng/mL. Serum digoxin levels should be interpreted with caution. In acute exposure, digoxin is absorbed into the plasma and then slowly redistributes into the tissues. As a result, high acute digoxin levels are not always associated with toxicity. Levels greater that 10 ng/mL at any time may be associated with significant toxicity, and treatment with digoxin-specific Fab should be considered. Other cardiac glycosides can cross-react with the digoxin assay, though the degree of this cross-reactivity varies. A false-positive assay may result in the presence of digoxin-like immunoreactive substance seen in neonates, pregnancy, renal insufficiency, liver disease, congestive heart failure, acromegaly, and stress.
Management. Cardiac glycoside toxicity should be considered in any patient taking digoxin and presenting with new-onset dysrhythmias, gastrointestinal complaints, or altered mental status. Meticulous attention to supportive care and a search for easily correctable conditions, such as hypoxia, hypovolemia, and electrolyte disturbances, are top priorities. All patients should be treated as discussed in the above section on "General Management of Cardiac Toxins."
In cardiac glycoside-poisoned patients, Fab fragments are the first-line therapy in patients with symptomatic cardiac dysrhythmias.41 Since cardiac glycoside-enhanced vagal activity is reversed by atropine sulfate, this agent has been used successfully in patients exhibiting AV block. Cardiac pacing has been advocated for bradydysrhythmias unresponsive to atropine, but care should be exercised as the pacing wire itself may induce ventricular fibrillation.
The classic treatment for ventricular dysrhythmias is phenytoin, as it increases the ventricular fibrillation threshold in the myocardium and enhances conduction through the AV node. Lidocaine has been advocated for treatment of ventricular dysrhythmias due to digitalis toxicity, although it does not affect AV nodal conduction. Amiodarone was reported to be effective in two cases refractory to other antiarrhythmics. Quinidine and procainamide are contraindicated in digitalis toxicity because they depress AV nodal conduction and may worsen cardiac toxicity. Electrical cardioversion of the digitalis-toxic patient must be performed with extreme caution and considered only as a last resort. A low energy setting (e.g., 10-25 W-sec) should be used and preparations made to treat potential ventricular fibrillation.
Potassium. Supplemental potassium may be beneficial in chronic digitalis toxicity when diuretic-induced hypokalemia is a factor. It should be given cautiously, as renal failure may be the cause of chronic digitalis toxicity. The acutely poisoned patient should not routinely receive potassium because hyperkalemia is common. Patients with acute cardiac glycoside poisoning who exhibit hyperkalemia may be treated with intravenous glucose, insulin, and SB or continuous inhaled beta agents such as albuterol (if no tachydysrhythmia or ectopy), although these therapies may be ineffective. The exchange resin sodium polystyrene sulfonate also should be considered. However, the increased serum potassium level reflects a change in potassium distribution and not an increase in total body potassium stores. Hence, use of agents such as exchange resins may lead to total body potassium depletion and subsequent problems once the digitalis toxicity has abated. There is a theoretical concern that treating cardiac glycoside-induced hyperkalemia with intravenous calcium may enhance digitalis cardiac toxicity and should be avoided.
Magnesium. Hypomagnesemia has been reported in a significant number of patients with chronic cardiac glycoside toxicity. Intravenous administration of magnesium has been shown to counteract ventricular irritability from digitalis toxicity.42 The recommended dose of magnesium for malignant ventricular dysrhythmias is 2-4 g (10-20 mL of a 20% solution) given intravenously over one minute. It should be infused more slowly in patients with ectopy who are hemodynamically stable. Magnesium also may be helpful in treating hyperkalemia. It should be used with extreme caution in renal failure patients.
Digoxin-Specific Fab. A milestone in the treatment of cardiac glycoside poisoning was the development of drug-specific antibodies. Digoxin-specific Fab fragments (Digibind or DigiTab) are antibody fragments produced by enzymatic cleavage of sheep immunoglobulin (IgG) antibodies to digoxin. Fab fragments can reverse digitalis-induced dysrhythmias, conduction disturbances, myocardial depression, and hyperkalemia in severely poisoned patients. Most patients have an initial response to cardiac glycoside toxic dysrhythmias within 30 minutes of Fab administration, and those who responded had complete resolution by four hours. Animal studies and case reports have demonstrated the efficacy of Fab fragments to the cardiac glycoside contained in plants. Adverse reactions to Fab administration have been few and include rare but mild hypersensitivity reactions, precipitous drops in serum potassium, and supraventricular tachydysrhythmias previously controlled by digoxin.
Fab fragment therapy should be administered for the following indications: 1) potassium greater than 5.0 mEq/L following acute ingestion; 2) serum digoxin concentration greater than 10 ng/mL; and 3) patients with potentially life-threatening dysrhythmias. Often, chronically poisoned patients can be managed by discontinuing digoxin and closely observing them. However, the threshold for treatment with Fab should be lower in chronically poisoned patients with signs of cardiac toxicity or who have chronic pulmonary disease, hypokalemia, hypothyroidism, renal insufficiency, or underlying cardiac disease.43 If patients are managed conservatively, the Fab dose to be administered should be calculated and the Fab fragments made available at the bedside while the patient is monitored for worsening toxicity.
Although serum digoxin levels should not be the sole factor in determining the need to administer Fab, dosage calculations for Fab are based on the serum digoxin level or estimated body load of digoxin. It is assumed that equimolar doses of antibody fragments are required to achieve neutralization. Forty milligrams of Fab (one vial) will bind 0.6 mg of digoxin. A severely toxic patient in whom the quantity ingested acutely is unknown should be given 5-10 vials at a time, and the clinical response should be observed. If cardiac arrest is imminent or has occurred, the dose can be given as a bolus. Otherwise, it should be infused over 30 minutes. In contrast, patients with chronic therapeutic overdose often have only mildly elevated digoxin levels and respond to one to two vials of Fab. The recommended dose for a given patient can be determined using the tables in the package insert or by contacting a regional poison center or toxicology consultant.
Free digoxin levels are decreased to zero within one minute of Fab fragment administration, but total serum digoxin levels markedly are increased. Since most assay methods measure both bound and free digoxin (total), very high digoxin levels are seen after Fab fragment therapy, but they have no correlation with toxicity. Serum levels may be unreliable for several days after Fab treatment. The digoxin-Fab complex is excreted in the urine and in patients with renal failure, elimination of the digoxin-Fab complex is prolonged and free digoxin levels gradually increase over hours after Fab administration. Rebound cardiac glycoside toxicity is rare but has been reported. Hemodialysis does not enhance elimination of digoxin-Fab complex.
Disposition. All patients who receive Fab fragments require continued monitoring in an intensive care unit for at least 24 hours. For the chronically poisoned elderly patient, modifying the outpatient treatment regimen by discontinuing the use of a cardiac glycoside or providing a more reliable method of drug administration with close clinical follow-up may avert further toxic episodes.
Conclusion
There are numerous agents that can result in human cardiotoxicity. The management of the toxicity of each should follow a rational approach as outlined above.
References
1. Albertson TE, Dawson A, de Latorre F, et al. TOX-ACLS: Toxicologic-oriented advanced cardiac life support. Ann Emerg Med 2001;37:S78-S90.
2. Litovitz TL, Klein-Schwartz W, Rodgers GC, Jr, et al. 2001 Annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2002;20:391-452.
3. Pond SM, Lewis-Driver DJ, Williams GM, et al. Gastric emptying in acute overdose: A prospective randomised controlled trial. Med J Aust 1995;163: 345-349.
4. DeRoos F. Calcium Channel Blockers. In: Goldfrank L, ed. Goldfrank’s Toxicologic Emergencies. 7th ed. New York: McGraw-Hill; 2002.
5. Holstege C, Kirk M, Furbee R. Wide complex dysrhythmia in calcium channel blocker overdose responsive to sodium bicarbonate therapy (abstract). J Toxicol Clin Toxicol 1998;36:509.
6. Kline JA, Raymond RM, Schroeder JD, et al. The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol 1997;145:357-362.
7. Tanen DA, Ruha AM, Curry SC, et al. Hypertonic sodium bicarbonate is effective in the acute management of verapamil toxicity in a swine model. Ann Emerg Med 2000;36:547-553.
8. Sami-Karti S, Ulusoy H, Yandi M, et al. Non-cardiogenic pulmonary oedema in the coarse of verapamil intoxication. Emerg Med J 2002;19:458-459.
9. Howarth DM, Dawson AH, Smith AJ, et al. Calcium channel blocking drug overdose: An Australian series. Hum Exp Toxicol 1994;13:161-166.
10. Isbister G. Delayed asystolic arrest after diltiazem overdose; resuscitation with high dose intravenous calcium. Emerg Med J 2002;19:355-357.
11. Papadopoulos J, O’Neil MG. Utilization of a glucagon infusion in the management of a massive nifedipine overdose. J Emerg Med 2000;18:453-455.
12. Snook CP, Sigvaldason K, Kristinsson J. Severe atenolol and diltiazem overdose. J Toxicol Clin Toxicol 2000;38:661-665.
13. Kline JA, Leonova E, Raymond RM. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med 1995;23:1251-1263.
14. Boyer EW, Shannon M. Treatment of calcium-channel-blocker intoxication with insulin infusion. N Engl J Med 2001;344:1721-1722.
15. Boyer EW, Duic PA, Evans A. Hyperinsulinemia/euglycemia therapy for calcium channel blocker poisoning. Pediatr Emerg Care 2002;18:36-37.
16. Yuan TH, Kerns W, II, Tomaszewski CA, et al. Insulin-glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol 1999;37:463-474.
17. Love JN, Howell JM, Newsome JT, et al. The effect of sodium bicarbonate on propranolol-induced cardiovascular toxicity in a canine model. J Toxicol Clin Toxicol 2000;38:421-428.
18. Love JN, Enlow B, Howell JM, et al. Electrocardiographic changes associated with beta-blocker toxicity. Ann Emerg Med 2002;40:603-610.
19. Brubacher J. Beta-adrenergic blockers. In: Goldfrank L, ed. Goldfrank’s Toxicologic Emergencies. 7th ed. New York: McGraw-Hill; 2002.
20. White C. A review of potential cardiovascular uses of intravenous glucagon administration. J Clin Pharmacol 1999;39:442-447.
21. Love JN, Hanfling D, Howell JM. Hemodynamic effects of calcium chloride in a canine model of acute propranolol intoxication. Ann Emerg Med 1996; 28:1-6.
22. Kearns W, Schroeder D, Williams C, et al. Insulin improves survival in a canine model of acute beta-blocker toxicity. Ann Emerg Med 1997;29: 748-757.
23. Sato S, Tsuji MH, Okubo N, et al. Combined use of glucagon and milrinone may not be preferable for severe propranolol poisoning in the canine model. J Toxicol Clin Toxicol 1995;33:337-342.
24. Sato S, Tsuji MH, Okubo N, et al. Milrinone versus glucagon: Comparative hemodynamic effects in canine propranolol poisoning. J Toxicol Clin Toxicol 1994;32:277-289.
25. Love JN, Howell JM, Litovitz TL, et al. Acute beta blocker overdose: Factors associated with the development of cardiovascular morbidity. J Toxicol Clin Toxicol 2000;38:275-281.
26. Kolecki PF, Curry SC. Poisoning by sodium channel blocking agents. Crit Care Clin 1997;13:829-848.
27. Harrigan RA, Brady WJ. ECG abnormalities in tricyclic antidepressant ingestion. Am J Emerg Med 1999;17:387-393.
28. Brady WJ, Skiles J. Wide QRS complex tachycardia: ECG differential diagnosis. Am J Emerg Med 1999;17:376-381.
29. Kerr GW, McGuffie AC, Wilkie S. Tricyclic antidepressant overdose: A review. Emerg Med J 2001;18:236-241.
30. Sides GD. QT interval prolongation as a biomarker for torsades de pointes and sudden death in drug development. Dis Markers 2002;18:57-62.
31. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol 2002;40:867-879.
32. Munro P, Graham C. Torsade de pointes. Emerg Med J 2002;19:485-486.
33. Kyrmizakis DE, Chimona TS, Kanoupakis EM, et al. QT prolongation and torsades de pointes associated with concurrent use of cisapride and erythromycin. Am J Otolaryngol 2002;23:303-307.
34. Horowitz BZ, Bizovi K, Moreno R. Droperidol—Behind the black box warning. Acad Emerg Med 2002;9:615-618.
35. De Ponti F, Poluzzi E, Cavalli A, et al. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: An overview. Drug Saf 2002;25:263-286.
36. Priori SG, Cantu F, Schwartz PJ. The long QT syndrome: New diagnostic and therapeutic approach in the era of molecular biology. Schweiz Med Wochenschr 1996;126:1727-1731.
37. Anderson ME, Al-Khatib SM, Roden DM, et al. Cardiac repolarization: Current knowledge, critical gaps, and new approaches to drug development and patient management. Am Heart J 2002;144:769-781.
38. Kaye P, O’Sullivan I. The role of magnesium in the emergency department. Emerg Med J 2002;19:288-291.
39. Monraba R, Sala C. Percutaneous overdrive pacing in the out-of-hospital treatment of torsades de pointes. Ann Emerg Med 1999;33:356-357.
40. Eddleston M, Ariaratnam CA, Sjostrom L, et al. Acute yellow oleander (Thevetia peruviana) poisoning: Cardiac arrhythmias, electrolyte disturbances, and serum cardiac glycoside concentrations on presentation to hospital. Heart 2000;83:301-306.
41. Kirk M, Judge B. Digitalis poisoning. In: Irwin J, ed. Intensive Care Medicine. 5th ed. Baltimore: Lippincott Williams & Wilkins; 2003.
42. Reisdorff EJ, Clark MR, Walters BL. Acute digitalis poisoning: The role of intravenous magnesium sulfate. J Emerg Med 1986;4:463-469.
43. Taboulet P, Baud FJ, Bismuth C. Clinical features and management of digitalis poisoning—Rationale for immunotherapy. J Toxicol Clin Toxicol 1993; 31:247-260.
The diagnosis and management of patients with manifestations of drug-induced cardiotoxicity is challenging for even the most experienced emergency physician. The following report reviews the pathophysiology and clinical manifestations of cardiotoxins to provide the front-line practitioners with evidence-based protocols for managing patients with life-threatening toxicity.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.