The Insulin Resistance Syndrome — Syndrome X
Authors: Serge A. Jabbour, MD, and Jeffrey L. Miller, MD, Division of Endocrinology, Diabetes & Metabolic Diseases; Department of Medicine, Thomas Jefferson University Hospital; Philadelphia, Penn.
Editor’s Note—The quintet of abdominal obesity, hypertension, dyslipidemia, abnormal glucose metabolism, and coagulopathy has been called "syndrome X" or "metabolic syndrome."1,2 Some have referred to this grouping as the "insulin resistance syndrome," the "obesity dyslipidemia syndrome," and the "deadly quartet," the end result being atherosclerosis, which leads to coronary artery disease and its sequelae.3-5
Insulin resistance plays a major role in the pathogenesis of type 2 diabetes (DM2) and in the associated metabolic abnormalities, such as dyslipidemia and hypertension. Insulin resistance is strongly linked to obesity (mainly abdominal visceral obesity), which is growing in major proportions within the developed world, especially the United States.6
DM2 is caused by a dual defect: insulin resistance and b-cell failure. Insulin resistance is a state in which a given concentration of insulin is associated with a subnormal glucose response.7 Thus, the b-cells of the pancreas secrete increased amounts of insulin to maintain euglycemia. However, over time, functional defects in insulin secretion prevent the b-cells from maintaining high rates of insulin secretion, resulting in impaired glucose tolerance and eventually DM2.8 In addition to causing hyperglycemia in DM2, insulin resistance and compensatory hyperinsulinemia also play a role in other metabolic abnormalities culminating in atherosclerosis and its sequelae (discussed later).
During the past decade, the cardiovascular complications associated with DM2 have attracted increasing attention. In fact, macrovascular complications account for 80% of the mortality in patients with DM2.9 Although aggressive glycemic control can delay or prevent the microvascular complications of the disease, which may eventually lead to renal failure, blindness, and neuropathy, there are no concrete data linking glycemic control to macrovascular complications.10,11 The reason is that the cardiovascular disease that accompanies diabetes is multi-factorial, including obesity, dyslipidemia and hypertension, and impaired hemostasis, eventually culminating in atherosclerotic vascular disease. It has been estimated that atherosclerosis has been progressing for 10-20 years before the clinical onset of DM2.2-5 In 2001, the Adult Treatment Panel III (ATP III) of the National Cholesterol Education Program recognized diabetes as a cardiovascular risk equivalent; a new definition for the metabolic syndrome (or syndrome X) as a treatment target in patients at risk for heart disease also was established.12
This review article will focus on the clinical spectrum of the metabolic syndrome, including obesity, abnormal glucose metabolism, hypertension, dyslipidemia, impaired hemostasis, macrovascular disease, and other associated features. The pathogenesis of each component in relation to insulin resistance and atherosclerosis and a brief overview of the therapeutic implications will be discussed, stressing appropriate early aggressive therapy of all risk factors.
Introduction
The metabolic syndrome is observed in at least 25% of adults 20-70 years of age in the United States,13 and occurs with increasing frequency in individuals with impaired glucose tolerance (approximately 45-65%) and DM2 (approximately 75-85%).14 The prevalence of the metabolic syndrome has an age-dependent increase (6.7% for ages 20-29, as opposed to 42% for ages > 70 years).13 Mexican-Americans had the highest age-adjusted prevalence (31.9%). Among African-Americans and Mexican-Americans, the prevalence was higher in women than in men (57% and 26% higher prevalence, respectively). The high prevalence of this syndrome and its strong association with obesity highlights the importance of appropriate lifestyle intervention (diet and exercise).
The ATP III tabulates 5 clinically determined characteristics of the metabolic syndrome and notes that the presence of any 3 is sufficient to confirm the diagnosis (see Table).12 Other components of the metabolic syndrome might include impaired hemostasis, cardiovascular disease, cutaneous abnormalities, hepatic steatosis, hyperandrogenism and reproductive abnormalities.6

Clinical Spectrum
Obesity
In the United States, the prevalence of obesity has reached epidemic proportions in the past decade. Obese patients are often insulin-resistant and hyperinsulinemic, and these 3 factors are characteristic of the prediabetic phase. Obesity, in particular increased adiposity in the visceral compartment (ie, central or abdominal obesity), is negatively correlated with insulin sensitivity.15 There are several mechanisms to explain how obesity, especially visceral obesity, leads to insulin resistance and contributes to cardiovascular disease. Visceral adipocytes are far more metabolically active than subcutaneous adipocytes. Lipolysis (ie, release of free fatty acids) from visceral fat is more pronounced than from subcutaneous fat, and visceral fat cells are less sensitive to suppression of lipolysis by insulin.16 The released free fatty acids can directly block insulin-signaling pathways,17 leading to insulin resistance. In addition, visceral fat has direct access to the portal circulation. Increased amounts of free fatty acids being released into the portal circulation may impair the metabolism and action of insulin and increase gluconeogenesis in the liver.18 Also, different mediators secreted by the adipocytes have been implicated in the pathogenesis of insulin resistance associated with obesity. These mediators or chemical messengers, collectively known as "adipocytokines," include tumor necrosis factor (TNF)-a, adiponectin, resistin, and leptin (see Figure 1).19,20
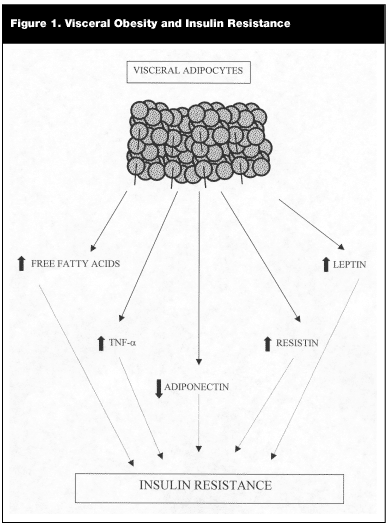
TNF-a is overexpressed in the adipose tissue of obese humans, and increases free fatty acid secretion.21 Adiponectin appears to enhance insulin action; as an individual becomes more obese, the adiponectin concentration in the blood is decreased and insulin resistance increases.22 Resistin is another recently identified adipocyte-specific protein found in mouse cells.23 Resistin is associated with insulin resistance and work to date has shown that resistin itself appears to be limited in expression to rodents, but there is a family of related molecules expressed in human fat tissue that may have effects similar to those of resistin.24 Treatment of rodents with rosiglitazone results in decrease in resistin and resultant decrease in insulin resistance.23 Leptin is another adipose-specific hormone that contributes to appetite regulation and has been proposed to affect insulin sensitivity. Some investigators have suggested that hyperleptinemia plays a crucial role in insulin resistance.25
Finally, aging is also associated with a higher prevalence of type 2 diabetes. This can be attributed to loss of lean body mass and an increase in adipose tissue, especially in sedentary individuals. This results in less muscle tissue available for glucose disposal and relatively more adipose tissue, leading to insulin resistance in susceptible individuals.
Visceral obesity, besides promoting the development of insulin resistance and DM2, is also an independent significant predictor of coronary heart disease morbidity and mortality.15,26
Hyperglycemia
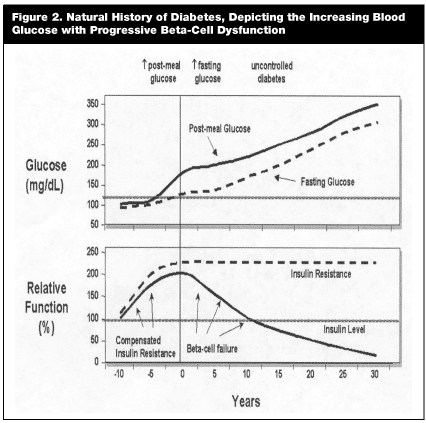
In order to maintain normal glucose levels in the blood circulation, insulin must be available and capable through its cellular action to promote the uptake and metabolism (disposal) of glucose into skeletal muscle and adipose tissue. In addition, sufficient insulin action must occur in the liver to suppress hepatic glucose production, the major source of circulating glucose in the fasting state.27 Several pathophysiologic changes have been shown to precede the development of the hyperglycemia of DM2.18 One essential factor is the appearance of insulin resistance, such as seen in patients with metabolic syndrome, impairing the use of glucose by peripheral tissues. Insulin resistance can be worsened by a variety of factors, including increases in body mass due to excess adiposity, sedentary lifestyle, age, and glucocorticoids. To compensate, the pancreas attempts to overcome the underlying defect of insulin resistance by increasing insulin secretion with subsequent hyperinsulinemia.8 If b-cells are normal, people remain euglycemic; but in the presence of functional defects in glucose-stimulated insulin secretion in the b-cells (mostly genetic in origin), hyperglycemia ensues.18,28 The compensated state of insulin resistance and hyperinsulinemia may last for a decade or more, but ultimately, insulin secretion decreases because of b-cell failure (see Figure 2).
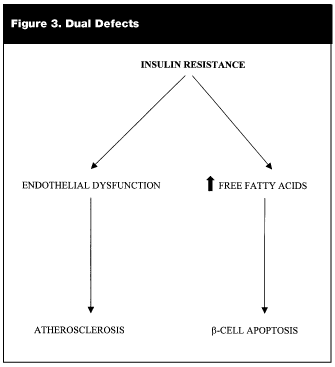
Free fatty acids (released from visceral fat) not only seem to interfere with insulin action but also with insulin secretion.28a It has been suggested that alterations in the expression of metabolic enzymes by free fatty acids may account for b-cell insensitivity to glucose or for alterations in insulin secretion.28a At least part of the reason behind b-cell failure is insulin resistance leading to elevated free fatty acids which result in b-cell apoptosis (see Figure 3).
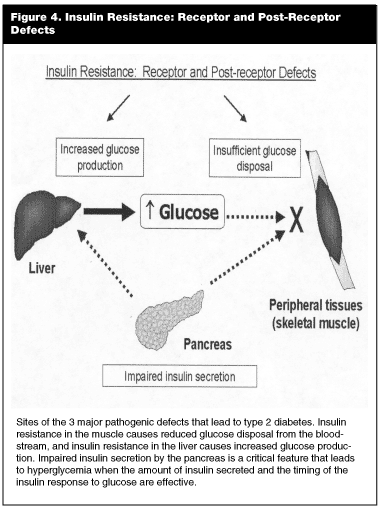
Hyperglycemia and DM2 develop when these 2 defects, insulin resistance and impaired b-cell function, are present.18 An early defect is a relative insulin deficiency leading to an increase in postmeal glucose levels, due to decrease in glucose use. A later effect is the increase in glucose production by the liver, especially in the fasting state, which is due to insufficient insulin action in the liver. Thus, persons with DM2 have 3 major pathophysiologic features that provide a useful framework on which to design the basis of a "stepped" therapeutic approach: insulin resistance with defective glucose disposal after meals in muscle and fat tissue, insulin resistance with increased glucose output by the liver and impaired b-cell function27 (see Figure 4). During the development of DM2, b-cell function is progressively lost. At the time of the diagnosis of diabetes, up to 50% of b-cell function has already been lost, and declines progressively over time (4% per annum).29 The degree of b-cell function is critical, because therapeutic approaches to prevent or treat diabetes are more effective earlier in the disease, most likely because b-cell response is more robust.6,29
Hypertension
Insulin resistance and subsequent hyperinsulinemia cause exaggerated responses in tissues that remain sensitive to insulin, underlying many of the pathophysiologic features of the insulin resistance syndrome, such as hypertension. Insulin resistance and hypertension are associated, although not as strongly as insulin resistance and dyslipidemia. Patients with DM2 are almost twice as likely to have hypertension as patients without diabetes, and approximately 50% of patients with hypertension are insulin-resistant and hyperinsulinemic.4 The rise in plasma insulin levels may elevate the blood pressure by one or more of the following mechanisms:4,5
• Hyperinsulinemia acts centrally to enhance the activity of the sympathetic nervous system.4,30 This hyperadrenergic state stimulates thermogenesis, thereby minimizing further weight gain. However, the price paid for this maintenance of energy balance is a sympathetically induced rise in the systemic blood pressure.
• Insulin and increased sympathetic activity can stimulate renal sodium reabsorption,3,5,31,32 leading to volume expansion, and elevation in systemic blood pressure.
• There may be a difference in the action of insulin on the vasculature. Insulin normally causes vasodilatation and enhanced muscle blood flow, an effect that appears to be mediated in part by nitric oxide; this effect is regulated by an enzyme involved in glucose metabolism.33,34 These effects are blunted in both obese and hypertensive subjects.33 Insulin resistance may result in proportionate reductions in glucose use and nitric oxide production (and therefore vasodilation).35
Despite all of these suggestive findings, there are several observations that do not confirm a tight causal relationship between insulin and hypertension:32
• Some epidemiologic studies have not been able to demonstrate a strong correlation between hyperinsulinemia and hypertension.36
• High insulin levels alone appear to be insufficient to substantially raise the blood pressure. As an example, chronic hyperinsulinemia (by infusion) does not induce hypertension in dogs.37 Similarly, humans with insulinoma do not become hypertensive and their blood pressure does not fall after successful surgery.38
These negative findings suggest that, if insulin is important in the pathogenesis of hypertension, there is substantial interpatient variability and that some additional factor or factors must also be present.30,39 One possibility is genetic susceptibility to the development of hypertension or to the effects of insulin on blood pressure.
Dyslipidemia
A central component of the insulin resistance syndrome is a characteristic pattern of lipid abnormalities, including elevated plasma triglyceride concentrations and decreased high-density lipoprotein (HDL) cholesterol. Although plasma concentrations of low-density lipoprotein (LDL) cholesterol may not differ from those in normal subjects, there is an increase in small, dense LDL particles in patients with insulin resistance.40,41 Elevated triglycerides, low levels of HDL and the presence of small, dense LDL are each independent risk factors for cardiovascular disease,12,42,43 and represent a set of lipoprotein abnormalities that promote atherosclerosis.44
Dyslipidemia associated with insulin resistance is thought to be initiated by the resistance of adipose cells to the effects of insulin. The inability of insulin-resistant fat cells to store triglycerides results in hydrolysis of triglycerides and release of fatty acids.45 The resulting availability of free fatty acids to the liver leads to increased hepatic synthesis and release of triglycerides and very low-dense lipoprotein (VLDL) cholesterol. Exchange of cholesterol esters from HDL and LDL to VLDL for triglyceride molecules renders HDL unavailable to remove cholesterol from peripheral cells. Thus, hypertriglyceridemia leads to low levels of HDL cholesterol. Furthermore, triglyceride-enriched LDL is readily converted to small, dense LDL, which has greater atherogenic potential, because it is more readily oxidized. Oxidized LDL is more readily scavenged by vascular scavenger LDL receptors and has a longer residence time in the vascular matrix. This leads to enhanced lipid deposition in the arterial wall. Oxidized LDL cholesterol also is toxic to endothelial cells, leading to decreased nitric oxide release and enhanced expression of cytokines and adhesion molecules. These effects in turn lead to vascular inflammation.46
The presence of small, dense LDL cholesterol has been associated with worsened cardiovascular outcomes. In the Quebec Cardiovascular Study, 2103 men without ischemic heart disease were followed during a period of 5 years.43 LDL particle size was measured in 103 patients who developed ischemic heart disease to determine the relationship between ischemic heart disease and presence of small, dense LDL. Notably, small, dense LDL composition, as described in the metabolic syndrome, was associated with a 3.6-fold increase in the risk of developing ischemic heart disease.
Individuals who are insulin-resistant or have DM2 uncommonly have elevated total LDL cholesterol levels,45 which suggests that the increased atherogenicity associated with diabetes is related to specific aspects of the LDL cholesterol lipid profile. The elevation in small, dense LDL composition is associated with increased triglycerides and reduced HDL cholesterol, all of which are risk factors for development of heart disease.43,45,46
Impaired Hemostasis
A more recently recognized component of the insulin resistance syndrome is a prothrombotic state. Patients with insulin resistance often have evidence of alterations in coagulation that predispose to arterial thrombosis.44,47 One of these alterations is increased levels of plasminogen activator inhibitor type 1 (PAI-1).
Maintenance of hemostasis is mediated through such factors as PAI-1 and tissue plasminogen activator (tPA). tPA mediates clot lysis by activating the fibrinolytic system. PAI-1 blocks the action of tPA and is the primary inhibitor of endogenous fibrinolysis.40 Elevated PAI-1 plasma concentrations are associated with cardiovascular disease and appear to be important in the pathogenesis of thrombosis and myocardial infarction. In persons with type 2 diabetes and in nondiabetic persons with insulin resistance, PAI-1 concentrations are elevated. There is a significant direct correlation between PAI-1 concentrations and insulin resistance.41 There are also strong, positive associations between fasting insulin concentrations and other markers of hemostatic function (eg, von Willebrand factor antigen, factor VII antigen, fibrinogen, and plasma viscosity).47
Thus, insulin resistance and hyperinsulinemia may increase the risk of cardiovascular disease by impairing hemostatic function and enhancing the potential for acute thrombosis.
Macrovascular Disease
Although many of the individual components of the metabolic syndrome predict increased risk for cardiovascular disease, presence of the metabolic syndrome is clearly associated with a high relative risk for coronary artery disease (CAD) (2.96), myocardial infarction (2.63), and stroke (2.27). This risk is greater than the risk associated with any of the individual components of the metabolic syndrome. For example, the relative risks for CAD for obesity, hypertension or dyslipidemia are 1.44, 1.57, and 1.73, respectively.48,49
The fact that individuals with metabolic syndrome have a markedly increased risk of CAD was also illustrated in a prospective epidemiologic study of 970 men with no CAD who were followed for 22 years; the presence of hyperinsulinemia was associated with an increased risk of a major coronary event, death or nonfatal myocardial infarction, although its predictive value diminished with time.50 The hazard ratios, adjusted for other risk factors, at 5, 10, 15, and 22 years were 2.3, 2.4, 1.8, and 1.3, respectively. Hyperinsulinemia was associated with increases in both cardiovascular and noncardiovascular mortality.51 Of note, there are little data supporting insulin itself as being atherogenic. The likely culprit might be some other atherogenic factors (? pro-insulin) secreted by the b-cells or the other components of the metabolic syndrome (dyslipidemia, hypertension. . .). In fact, aggressive insulin therapy may lead to a reduction in mortality from cardiovascular disease, as shown in the Diabetes Mellitus Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) trial.51a
Impaired glucose tolerance and DM2 are associated with increased risk for cardiovascular morbidity and mortality. The increased risk of macrovascular complications likely begins years before the development of clinical type 2 diabetes, when insulin resistance and hyperinsulinemia are present. Prediabetic subjects usually have hyperinsulinemia and a more atherogenic pattern of cardiovascular risk factors compared with subjects who do not develop diabetes.52
Atherosclerosis is now recognized as an inflammation of the arterial wall.52a It starts with abnormalities in the endothelium (from hypertension, dyslipidemia, diabetes and other factors), resulting in the adhesion of circulating monocytes and T-cells, and in the formation of an inflammatory nidus. The adherent monocytes are then activated and move into the subendothelial space, where they form "foam cells" loaded with oxidized low-density lipoprotein. These foam cells are active and secrete matrix metalloproteinase, which may lyse the fibrous cap of the atherosclerotic plaque to make the plaque unstable and to cause plaque rupture. Such a rupture leads to initiation of the thrombotic process.
After individuals have developed subclinical or clinical cardiovascular disease, the presence of the disease is far more predictive of future cardiovascular events than the components of the metabolic syndrome.53 Therefore, it may be concluded that treatment of the metabolic syndrome is likely to maximally reduce future development of cardiovascular disease, if treatment begins before significant structural vascular damage has occurred. The common soil hypothesis linking DM2, hypertension, dyslipidemia, and atherosclerosis is inflammation.
Other Associated Features
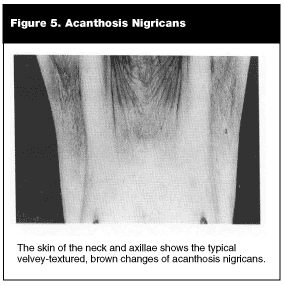
Acanthosis nigricans and skin tags are commonly associated with primary insulin resistance, regardless of its molecular cause. Acanthosis nigricans is a skin lesion characterized by brown, velvety, hyperkeratotic plaques54 (see Figure 5). The lesions are usually found on the back of the neck, the axilla, the groin, and over the elbows, but they may cover the entire surface of the skin, sparing only the palms and soles. The common denominator in all cases of acanthosis nigricans, with the possible exception of tumor-induced lesions, is insulin resistance.
Men with insulin resistance are not known to have disorders of the reproductive system. In contrast, women with insulin resistance commonly present with reproductive abnormalities.55 Insulin resistance may lead to ovarian hyperandrogenism, a sequence that has been proposed in women with polycystic ovary syndrome (PCOS), where women present with hirsutism and irregular menses. Of course, hyperinsulinemia by itself is not enough to induce PCOS, which may arise as a complex genetic disorder in which an intrinsic ovarian genetic trait interacts with other congenital or cellular environmental factors (like insulin resistance) to cause dysregulation of steroidogenesis.56
The metabolic syndrome also has been associated with abnormal liver pathology in very obese patients including steatosis, fibrosis, and cirrhosis.57
Therapeutic Implications
Because syndrome X is associated with significant morbidity and mortality, early diagnosis and aggressive treatment are critical, especially in DM2, where complications often are present years before diagnosis. Adverse sequelae are easier to prevent than to reverse, and preservation of b-cell function may slow progression of the disease.
Weight Loss
Regardless of the mechanisms, avoidance of obesity, particularly abdominal obesity, should reduce the potential for the development of the various features of the insulin resistance syndrome. For most patients, weight reduction and increased physical activity will improve insulin resistance, hyperglycemia, hypertension, and dyslipidemia, and reduce the risk of cardiovascular disease.
Recently, the Diabetes Prevention Program Research Group58 has shown that lifestyle changes can slow the conversion of pre-diabetes to DM2. This study randomized 3234 patients with impaired glucose tolerance to treatment with placebo, metformin (850 mg twice a day), or lifestyle modification (goal of at least 7% weight loss and at least 150 minutes of physical activity per week). Patients were monitored for a period of 2.8 years. Compared with placebo, patients who modified their lifestyle experienced a 58% reduction in the incidence of diabetes and patients treated with metformin had a 31% reduction.
Similarly, another study from Finland59 randomized 522 overweight patients with impaired glucose tolerance to lifestyle modification (goal of at least 5% weight loss and moderate exercise for at least 30 minutes per day) or control group. After a follow-up of 3.2 years, the risk of diabetes was reduced by 58% in the intervention group.
These data suggest that lifestyle modification is extremely effective for preventing or delaying the onset of diabetes.
When present, DM2 is a disease associated with significant morbidity and mortality; so early diagnosis and aggressive treatment are critical. Early intervention in patients with DM2 is particularly important, because complications often are present before diagnosis, adverse sequelae are easier to prevent than to reverse, and preservation of b-cell function may slow progression of the disease. This is why treating the pre-diabetes state is paramount, primarily through weight loss and targeting the other cardiovascular risk factors.
For patients with DM2, specific treatment goals have been established:62,63
- HbA1c < 7% (American Diabetes Association);
- HbA1c < 6.5% (American College of Endocrinology);
- Blood pressure < 130/80; and
- LDL < 100 mg/dL.
Glycemic Control
The UK Prospective Diabetes Study (UKPDS) established that treatment of patients with DM2 with sulfonylureas, metformin, or insulin reduced the development of microvascular complications, including retinopathy and nephropathy.11,60 In patients receiving intensive therapy with either a sulfonylurea or insulin, the microvascular complication rate was reduced by 25% compared with patients who received conventional dietary therapy.11 In overweight patients, metformin decreased the incidence of myocardial infarction by almost 39%. Sulfonylurea, metformin, and insulin therapies were similarly effective in improving glucose control; each therapeutic agent, as monotherapy, increased 2- to 3-fold the proportion of patients who attained HbA1c below 7% compared with diet alone.61 However, the progressive deterioration of diabetes control was such that after 3 years, approximately 50% of patients could attain this goal with either monotherapy (sulfonylurea, metformin, or insulin), and by 9 years this declined to approximately 25%.61 The big lesson we learned from the UKPDS was that the majority of patients needed multiple combination therapies to attain the glycemic target levels in the long term.
Insulin Resistance
The thiazolidinediones (TZDs) directly improve insulin resistance. Because their antihyperglycemic effect is less pronounced than metformin or sulfonylureas as monotherapy, their use in combination therapy is favored, preferably early in the treatment of DM2; because TZDs have been shown to relocate visceral to subcutaneous fat and have anti-inflammatory properties in the vascular endothelium, both of which are anticipated to be beneficial in reducing atherosclerotic heart disease.41
TZDs have been shown to reduce PAI-1 levels and decrease C-reactive protein levels. Also, they appear to reduce the intima-media thickness of the carotid arteries.63a The TZD troglitazone also has been shown to be effective in preventing DM2 in a high-risk group of women who recovered from gestational diabetes (Troglitazone in the Prevention of Diabetes study [TRIPOD]),64 but no outcome data on other TZDs are available yet. In animal models, rosiglitazone, in addition to its anti-inflammatory properties, reduces free fatty acids (preventing b-cell apoptosis from high level of free fatty acids) and causes b-cell rejuvenation, explaining 3-3.5 year data with sustained b-cell efficacy.
In the UKPDS, the data demonstrated that for every 1% decrease in mean HbA1c levels, the risk of microvascular complications decreased by 37%, but the risk of myocardial infarction decreased by only 14%.65 The reason why macrovascular complications were not very significantly affected by glycemic control is probably due to the presence of other cardiovascular risk factors including hypertension and dyslipidemia.
Hypertension
There is evidence that treatment with angiotensin-converting enzyme inhibitors exerts a vascular-protective effect and improves cardiovascular outcomes in high-risk or diabetic patients.66,67 The Heart Outcomes Prevention Evaluation (HOPE) study evaluated the effects of ramipril on cardiovascular events in patients at high risk for cardiovascular disease (9297 patients, including 3577 with diabetes). After 5 years, treatment with ramipril reduced the rates of death from cardiovascular causes by 26%, myocardial infarction by 20%, and stroke by 32%; more importantly, ramipril reduced the risk of new cases of diabetes by 34%, which may be due to a differential effect on insulin resistance. Almost similar reductions were seen in the subgroup of patients with diabetes, where the ramipril treatment group also had a 16% reduction in overt nephropathy, dialysis, or laser therapy. Only a small part of the benefit could be attributed to a reduction in blood pressure.
Another study examined the effect of an angiotensin-II receptor antagonist (losartan) in 9193 patients (including 1195 with diabetes) with hypertension and left ventricular hypertrophy (Losartan Intervention For Endpoint reduction trial [LIFE]).68 The trial was randomized against atenolol. After 4 years, losartan was better than atenolol in reducing the frequency of the primary composite end point of cardiovascular death, stroke, and myocardial infarction. Also, there was a lower rate of new-onset diabetes (difference of 25%) with losartan.
Dyslipidemia
As mentioned before, dyslipidemia is a major component of the metabolic syndrome and a risk factor for heart disease. It is well known from major clinical trials that lowering LDL to a target goal based on risk factors is beneficial and improves cardiovascular outcomes.69,70 When diabetes is present, the patient is considered as having a CAD risk equivalent12 and is at increased risk for developing subsequent cardiovascular complications. There is evidence to suggest that management of dyslipidemia in patients with diabetes can significantly reduce the incidence of both fatal and nonfatal coronary events.71,72 A subgroup of 202 patients with diabetes and established CAD was evaluated in the Scandinavian Simvastatin Survival Study (4S).71 After treatment with simvastatin, patients with diabetes had a 55% risk reduction for a major CAD event compared with a 32% reduction in patients without diabetes. The Cholesterol And Recurrent Events (CARE) trial evaluated the effect of pravastatin compared with placebo in 586 patients with diabetes and history of CAD.72 Treatment with pravastatin reduced the relative risk of coronary events by 25% in patients with diabetes and 23% in patients without diabetes.
Statins have also been shown to be effective in primary prevention of CAD. The West of Scotland Coronary Prevention Study (WOSCOPS) was designed to evaluate the effect of pravastatin for 4 years on the rate of nonfatal and fatal myocardial infarction (MI) in 6595 men without history of CAD. There was almost 31% reduction in the incidence of nonfatal MI and death from cardiac events.72a
Also, statins may have actions other than lipid lowering (antiinflammatory effect). In the Heart Protection Study (HPS),72b 20,536 subjects were assigned to simvastatin or placebo. Entry criteria included a history of CAD, diabetes or hypertension. Even in patients who had baseline LDL < 116 mg/dL, there was 18% reduction in deaths from heart disease and 24% reduction in major cardiovascular events.
Also, besides targeting LDL as a primary goal, increasing HDL as a secondary target improves cardiovascular outcomes. The Veterans Affairs HDL-Cholesterol Intervention Trial (VA-HIT) recruited men who had a CAD event in the past and dyslipidemia profile that included HDL cholesterol of less than 40 mg/dL. Patients were randomized to treatment with placebo or gemfibrozil and were followed for 5.1 years.73 Treatment with gemfibrozil decreased triglycerides by 31% and increased HDL cholesterol by 6% but had no effect on LDL cholesterol. The gemfibrozil-treated individuals had a 22% risk reduction for the combined end point of death from CAD and nonfatal myocardial infarction compared with the placebo-treated group.
Coagulopathy
Finally, because of the procoagulant state seen in metabolic syndrome, administration of aspirin has been shown to be beneficial in patients with coronary heart disease or diabetes.62 The most plausible mechanism for the benefit of aspirin relates to its ability to irreversibly inhibit platelet dependent cyclooxygenase agreeability, thereby reducing the risk of thrombotic vascular events. Small amounts of aspirin (75-81 mg) have such a pronounced effect that higher doses appear to yield no additional benefit. Also, naproxen (a nonsteroidal anti-inflammatory drug) appears to share anti-platelet effects with aspirin.74
Conclusion
In the metabolic syndrome or syndrome X, the cornerstone for management is to reduce the underlying cause, mainly obesity, which is a growing epidemic in the United Sates. For most patients, weight reduction and increased physical activity will improve insulin resistance, hyperglycemia, hypertension, and dyslipidemia, and reduce the risk of cardiovascular disease. If weight loss is not achieved, specific therapies should be targeted against the different components of the syndrome for each individual patient.
As macrovascular (cardiovascular) disease is the major cause of morbidity and mortality, and as these complications are established well before development of type 2 diabetes, early aggressive treatment aimed at reduction of visceral adiposity, glucose lowering, attention to blood pressure, dyslipidemia and prothrombotic state are essential. As the common soil hypothesis between glycemia, hypertension, dyslipidemia and atherosclerosis (as occurs in the metabolic syndrome) is inflammation, early aggressive treatment with insulin sensitizers, statins, tissue-specific ACE inhibitors and aspirin favors these agents as specifically anti-inflammatory.
References
1. Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595-1607.
2. Reaven GM. Role of insulin resistance in human disease (syndrome X): An expanded definition. Annu Rev Med. 1993;44: 121-131.
3. DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173-194.
4. Reaven GM, et al. Hypertension and associated metabolic abnormalities—The role of insulin resistance and the sympathoadrenal system. N Engl J Med. 1996;334:374-381.
5. Williams B. Insulin resistance: The shape of things to come. Lancet. 1994;344:521-524.
6. Goldstein BJ. Insulin resistance as the core defect in type 2 diabetes mellitus. Am J Cardiol. 2002;90(suppl 5A):3G-10G.
7. Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev. 1995;75:473-486.
8. DeFronzo RA, et al. Pathogenesis of NIDDM: A balanced overview. Diabetes Care. 1992;15:318-368.
9. Gu K, et al. Diabetes and decline in heart disease mortality in US adults. JAMA. 1999;281:1291-1297.
10. Diabetes Control and Complications Trial (DCCT) Research Group. Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial. Am J Cardiol. 1995;75:894-903.
11. UK Prospective Diabetes Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837-853.
12. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001; 285:2486-2497.
13. Ford ES, et al. Prevalence of the metabolic syndrome among US adults: Findings from the Third National Health and Nutrition Examination Survey. JAMA. 2002;287:356-359.
14. Isomaa B, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24: 683-689.
15. Carey DG, et al. Abdominal fat and insulin resistance in normal and overweight women: direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes. 1996;45:633-638.
16. Zierath JR, et al. Regional difference in insulin inhibition of non-esterified fatty acid release from human adipocytes: Relation to insulin receptor phosphorylation and intracellular signaling through the insulin receptor substrate-1 pathway. Diabetologia. 1998;41:1343-1354.
17. Dresner A, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253-259.
18. Lebovitz HE. Type 2 diabetes: An overview. Clin Chem. 1999; 45:1339-1345.
19. Montague CT, O’Rahilly S. The perils of portliness: Causes and consequences of visceral obesity. Diabetes. 2000;49:883-888.
20. Yamauchi T, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941-946.
21. Hotamisligil GS. Mechanisms of TNF-a-induced insulin resistance. Exp Clin Endocrinol Diabetes. 1999;107:119-125.
22. Matsuzawa Y, et al. Molecular mechanisms of metabolic syndrome X: Contributions of adipocytokines. Ann NY Acad Sci. 1999;892:146-154.
23. Steppan CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307-312.
24. Steppan CM, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA. 2001;98:502-506.
25. DeCourten M, et al. Hyperleptinaemia: The missing link in the metabolic syndrome? Diabet Med. 1997;14:200-208.
26. Fontbonne AM, Eschwege EM. Insulin and cardiovascular disease: Paris Prospective Study. Diabetes Care. 1991;14:461-469.
27. Jabbour SA, Goldstein BJ. Improving disease management with new treatments for type 2 diabetes mellitus. Clinical Geriatrics (special issue). 2001;9:57-68.
28. Kahn BB. Type 2 diabetes: When insulin secretion fails to compensate for insulin resistance. Cell. 1998;92:593-596.
28a. Stumvoll M, Gerich J. Clinical features of insulin resistance and beta cell dysfunction and the relationship to type 2 diabetes. Clin Lab Med. 2001;21:31-51.
29. Lebovitz HE. Insulin secretagogues: Old and new. Diabetes Rev. 1999;7:139-153.
30. Moan A, et al. Insulin sensitivity, sympathetic activity, and cardiovascular reactivity in young men. Am J Hypertens. 1995;8: 268-275.
31. Bigazzi R, et al. Clustering of cardiovascular risk factors in salt-sensitive patients with essential hypertension: Role of insulin. Am J Hypertens. 1995;9:24-32.
32. Hall JE. Renal and cardiovascular mechanisms of obesity. Hypertension. 1994;23:381-394.
33. Steinberg HO, et al. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601-2610.
34. Baron AD, et al. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J Clin Invest. 1995;96:786-792.
35. Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894-898.
36. Muller DC, et al. An epidemiological test of the hyperinsulinemia-hypertension hypothesis. J Clin Endocrinol Metab. 1993; 76:544-548.
37. Hall JE, et al. Hemodynamic and renal responses to chronic hyperinsulinemia in obese insulin-resistant dogs. Hypertension. 1995;25:994-1002.
38. O’Brien T, et al. Hypertension and dyslipidemia in patients with insulinoma. Mayo Clin Proc. 1993;68:141-146.
39. Saad MF, et al. Racial differences in the relation between blood pressure and insulin resistance. N Engl J Med. 1991;324:733-739.
40. Davidson MB. Clinical implications of insulin resistance syndrome. Am J Med. 1995;99:420-426.
41. Parulkar AA, et al. Nonhypoglycemic effects of thiazolidinediones. Ann Intern Med. 2001;134:61-71.
42. Fontbonne A, et al. Hypertriglyceridemia as a risk factor of coronary heart disease mortality in subjects with impaired glucose tolerance or diabetes: Results from the 11-year follow-up of the Paris Prospective Study. Diabetologia. 1989;32:300-304.
43. Lamarche B, et al. Small, dense low-density lipoprotein particles as a predictor of the risk of ischemic heart disease in men: Prospective results from the Quebec Cardiovascular Study. Circulation. 1997;95:69-75.
44. Grundy SM, et al. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation. 1999;100:1134-1146.
45. Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106:453-458.
46. Reusch JEB. Current concepts in insulin resistance, type 2 diabetes mellitus, and the metabolic syndrome. Am J Cardiol. 2002;90(suppl 5A):19G-26G.
47. Meigs JB, et al. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: the Framingham offspring study. JAMA. 2000; 283:221-228.
48. Isomaa B, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24: 683-689.
49. Lebovitz HE. Rationale for and role of thiazolidinediones in type 2 diabetes mellitus. Am J Cardiol. 2002;90(suppl):34G-41G.
50. Pyorala M, Miettinen H, Laakso M, et al. Hyperinsulinemia predicts coronary heart disease risk in healthy middle-aged men: The 22-year follow-up results of the Helsinki Policemen Study. Circulation. 1998;98:398-404.
51. Pyorala M, et al. Plasma insulin and all-cause, cardiovascular and noncardiovascular mortality: The 22-year follow-up results of the Helsinki Policemen Study. Diabetes Care. 2000;23:1097-1102.
51a. Malmberg K, et al. Effects of insulin treatment on cause-specific one-year mortality and morbidity in diabetic patients with acute myocardial infarction. Eur Heart J. 1996;17:1337-1344.
52. Haffner SM, et al. Cardiovascular risk factors in confirmed prediabetic individuals: does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA. 1990; 263:2893-2898.
52a. Ross R. Atherosclerosis: An inflammatory disease. N Engl J Med. 1999;340:115-126.
53. Kuller LH, et al. Diabetes mellitus: Subclinical cardiovascular disease and risk of incident cardiovascular disease and all-cause mortality. Arterioscler Thromb Vasc Biol. 2000;20:823-829.
54. Jabbour SA, Miller JL. Endocrinopathies and the skin. Int J Dermatol. 2000;39:88-99.
55. Poretsky L, Piper B. Insulin resistance, hypersecretion of LH, and a dual-defect hypothesis for the pathogenesis of polycystic ovary syndrome. Obstet Gynecol. 1994;84:613-621.
56. Rosenfield RL. Polycystic ovary syndrome and insulin-resistant hyperinsulinemia. J Am Acad Dermatol. 2001;45:S95-S104.
57. Marceau P, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab. 1999;84:1513-1517.
58. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393-403.
59. Tuomilehto J. et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343-1350.
60. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352:854-865.
61. UK Prospective Diabetes Study Group. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus. JAMA. 1999;281:2005-2012.
62. American Diabetes Association. Standards of medical care for patients with diabetes mellitus. Diabetes Care. 2002;25(suppl 1): S33-S49.
63. Feld S. The American Association of Clinical Endocrinologists medical guidelines for the management of diabetes mellitus: The AACE system of intensive diabetes self-management. Endocr Pract. 2002;8(suppl 1):40-82.
63a. Koshiyama H, et al. Inhibitory effect of pioglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab. 2001;86:3452-3456.
64. Buchanan TA, et al. Protection from type 2 diabetes persists in the TRIPOD cohort eight months after stopping troglitazone (abstract). Diabetes. 2001;50(suppl 2):A81.
65. UK Prospective Diabetes Study (UKPDS) Group. Association of glycaemia with macrovascular and microvascular complications in type 2 diabetes (UKPDS 35): Prospective observational study. BMJ. 2000;321:405-412.
66. The Heart Outcomes Prevention Evaluation (HOPE) Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular outcomes in high-risk patients. N Engl J Med. 2000;342:145-153.
67. Heart Outcomes Prevention Evaluation (HOPE) Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in patients with diabetes mellitus: Results of the HOPE and MICRO-HOPE substudy. Lancet. 2000;355:253-259.
68. Losartan Intervention For Endpoint reduction (LIFE) study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet. 2002; 359:1004-1010.
69. Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Survival Study (4S). Lancet. 1994; 344:1383-1389.
70. Cholesterol And Recurrent Events Trial Investigators. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996: 335:1001-1009.
71. Scandinavian Simvastatin Survival Study (4S) Group. Cholesterol lowering with simvastatin improves progression of diabetic patients with coronary heart disease: A substudy analysis of the Scandinavian Simvastatin Survival Study. Diabetes Care. 1997;20:614-620.
72. Goldberg RB, et al, for the CARE Investigators. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: Subgroup analysis in the Cholesterol And Recurrent Events (CARE) trial. Circulation. 1998;98: 2513-2519.
72a-Shepherd J, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301-1307.
72b. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360:7-22.
73. Rubins HB, et al, for the Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med. 1999;341:410-418.
74. Rahme E, et al. Association between naproxen use and protection against acute myocardial infarction. Arch Intern Med. 2002;162:1111-1115.
The quintet of abdominal obesity, hypertension, dyslipidemia, abnormal glucose metabolism, and coagulopathy has been called syndrome X or metabolic syndrome. Some have referred to this grouping as the insulin resistance syndrome, the obesity dyslipidemia syndrome, and the deadly quartet.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.