Hypermethylation of TPEF in Human Cancers
Hypermethylation of TPEF in Human Cancers
By Daniel J. Weisenberger, PhD, Peter A. Jones, PhD, DSc,and Gangning Liang, MD, PhD
Alterations in dna methylation patterns are a hallmark of human cancers. DNA methylation is prevalent at the C-5 position of cytosine in CpG dinucleotides.1 CpG sequences, although reduced in the genome due to an increase in the rate of spontaneous deamination of 5-methylcytosine over cytosine, usually are seen clustered in CpG islands.2 These are regions of greater than 200 bp DNA that have a G+C content greater than 0.5, and an observed/expected level of CpG greater than 0.6.3 CpG islands usually are found at the 5’ regions of genes, and extend from upstream of the transcripition initiation site into the exonic coding regions.4 CpG sequences within CpG islands are predominantly free of methylation, whereas CpG sequences outside of CpG islands show a high degree of methylation in somatic cells.2
Usually, methylation of CpG islands only has roles in X-chromosome inactivation and imprinting.5-7 Methylation of CpG islands in the promoter regions of genes is associated with transcription silencing;1,8,9 hypermethylation of the promoters of tumor-suppressor genes is believed to play a role in tumorigenesis, as there is substantial evidence of aberrant DNA methylation of tumor-suppressor genes in cancer.1,9,10 Therefore, the ability to identify hypermethylated regions of DNA provides a novel method of understanding the relationship of DNA methylation and cancer, and also the pathways of tumorigenesis in general.
The key to understanding the relationship between DNA methylation and cancer becomes an issue not only of understanding the mechanism of how DNA methylation contributes to tumorigenesis, but also of identifying such aberrant methylation patterns in the human genome. Recently, there have been numerous reports of genome scanning methods that can determine differential methylation patterns in cancer; those methods include genomic sequencing, Southern blotting, and methylation sensitive PCR analyses.11-13 We have used the Methylation-sensitive Arbitrarily-Primed PCR (Ms-AP-PCR) technique to rapidly screen for specific and unknown regions of DNA that are aberrantly methylated in tumors, and more importantly, to identify and discover new tumor-suppressor genes.14,15 In this method, genomic DNA isolated from tumors and normal tissues are digested with methylation-sensitive enzymes followed by PCR with random priming. Thus, different patterns of PCR products are generated, and PCR products that are differentially amplified based on their methylation status were isolated and sequenced. This technique has been quite effective in identifying tumor-suppressor genes that are differentially methylated in tumor tissues.
Hypermethylation of the TPEF Gene in Human Cancers
A recent study in our laboratory using the Ms-AP-PCR procedure has uncovered a DNA fragment that is hypermethylated in tumor samples.16 The sequence matched the 5’ region of a cDNA clone Expressed Sequence Tag (EST), implying that the fragment is located in the promoter or 5’ coding region of the gene. Determination of the gene’s transcription initiation site showed that the putative start site is only 12 bp upstream of the end of the EST sequence. The sequence of a clone of the gene containing the first exon revealed that the promoter was a CpG island and was TATA-less. A possible promoter region located from -650 to +1 was predicted after cloning and sequencing of this sequence. Sequencing data also indicate that the cDNA sequence of this gene (TPEF) is 1836 bp, and identifies a 1124 nucleotide open reading frame that encodes a 374 amino acid protein with a molecular weight of 41.4 kD and a translation start site that begins with ATG. Figure 1 shows the 5’ region of TPEF in which the promoter region is highlighted in gray.
| Figure 1. Hypermethylation of TPEF |
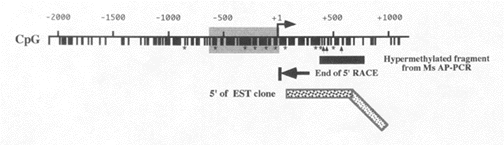 |
The predicted protein sequence also indicates signal peptide and transmembrane regions. BLAST searches using the protein sequence identified above showed a high degree of homology with 1) a transmembrane protein (TM) of Xenopus laevis with epidermal growth factor (EGF) and follistatin domains and 2) a human transmembrane protein. A comparison of sequence homology between these proteins shows approximately 50% homology of TPEF with the Xenopus TM protein and 72% homology to the human TM protein. Analysis of the TPEF gene indicates that it contains the EGF domain as well as two follistatin domains that are similar to those of the human and Xenopus TM proteins.17 Follistatin binds and inactivates activin and other members of the transforming growth factor (family of proteins.16,18 Therefore, TPEF may bind to these growth factors and suppress their actions. TPEF also is thought to act as a growth suppressor through its EGF domain. TPEF contains the EGF domain defined as CX7CX4-5 CX10-13CXCX8GXRC (where X is any amino acid other than C, G, or R). If the arginine (R) is mutated to a histidine in the EGF domain of human EGF, EGF no longer confers affinity for the EGF receptor. In TPEF, the arginine is absent, and thus TPEF may act as a ligand in inactivating the EGF receptor.16,19
Expression analysis using a human Northern master blot indicated that TPEF is expressed in a variety of
tissues, with a robust expression in prostate and brain and low levels of expression in other tissues. TPEF was localized to chromosome 2q33 by fluorescence in situ hybridization. Methylation levels of the 5’ region of TPEF also were measured using the Methylation-
sensitive Single Nucleotide Primer Extension (Ms-SNuPE) assay on bisulfite-treated genomic DNA.20 This procedure, developed in our laboratory, provides a quantitative measurement of methylation levels of specific CpG sites in DNA. Briefly, genomic DNA is treated with bisulfite to convert unmethylated cytosines to uracil (replicated as thymines), while 5-methylcytosine is left un-changed. The DNA region of interest is then amplified by PCR. Finally, primers are annealed to the PCR product and terminated immediately 5’ to the original CpG site of interest. Quantitation of the relative ratios of methylated vs. unmethylated cytosines (C or T) is determined by incubating this annealed product with Taq polymerase and either (a-32P) dCTP or (a-32P) dTTP, followed by gel electrophoresis and PhosphorImager analysis.
Ms-SNuPE analysis of methylation levels of the 5’ region of TPEF in a variety of bladder, colon, and prostate tumor cell lines revealed that this region of the gene (highlighted by the arrows in Figure 1) was hypermethylated in nine of the 11 tumor cell lines shown in Figure 2 (left panel). The hypermethylated cell lines exhibited values of DNA methylation that ranged from approximately 75% to 100%. RT-PCR analysis of these nine tumor cell lines also showed a loss of TPEF expression, whereas the two cell lines that did not exhibit TPEF hypermethylation showed expression of the gene. These results demonstrate that TPEF hypermethylation sufficiently blocks its expression.
| Figure 2. Methylation Levels of TPEF in Tumor Cell Lines and Primary Tumors | |
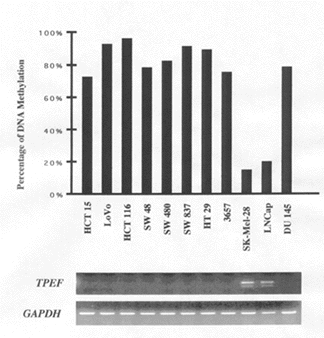 |
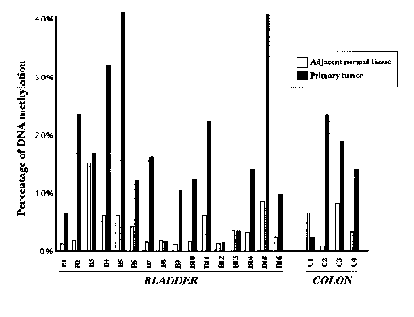 |
Ms-SNuPE analysis of TPEF hypermethylation in uncultured bladder and colon tumor tissues and adjacent normal tissues obtained from cancer patients (see Figure 2, right panel) showed that 11 of 16 bladder tumor tissue samples and three of four colon tumors were hypermethylated in the 5’ region of TPEF compared to the normal adjacent samples. Thus, 14 of 20 tumor samples showed TPEF hypermethylation, clearly demonstrating that TPEF hypermethylation is evident in tumors. Ms-SNuPE analysis, however, provides the average percent of methylation of a large number of individual molecules. To determine the TPEF methylation levels and patterns in individual molecules, the TPEF gene from a bladder cancer patient was cloned and colonies identified and sequenced. The sequencing analysis (see Figure 3) showed some TPEF methylation normally, with a significant increase of methylation in the tumor tissues. This suggests that TPEF methylation changes may be initiated in the pre-malignant tissues and accentuated in the tumor tissues.
| Figure 3. TPEF Sequencing Analysis |
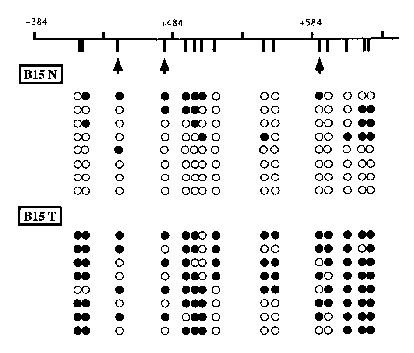 |
Conclusion
In the described study by Liang and colleagues, Ms-AP-PCR was used to identify a novel gene fragment that is hypermethylated in tumor tissues.16 The fragment matched the promoter region of the TPEF gene, a gene that encodes a protein that normally inhibits and suppresses cell growth. TPEF hypermethylation sufficiently inactivates its expression, thereby eradicating the anti-proliferative function of the translated protein. Here, we see a clear correlation between DNA hypermethylation and a tumorigenic phenotype. This is validated in separate experiments involving a distinct array of tumor cell lines as well as uncultured tumor tissue samples from cancer patients. Ms-SNuPE, as well as genomic sequencing, independently confirmed these results. Previously, the Ms-AP-PCR, as well as other methylation-sensitive genome scanning techniques, provided evidence for the involvement of DNA methylation in silencing genes that control cell growth and cell proliferation.
TPEF joins the growing list of genes found to be hypermethylated in tumor tissues, and further reinforces a role for DNA methylation in tumorigenesis. The extent by which hypermethylation is initiated is still not understood at the present time; however, the identification of genes that are hypermethylated in tumors does provide evidence that tumorigenesis can be initiated through a number of pathways that involve the loss of suppression of cell growth. The finding that TPEF exhibits regions of methylation in the normal adjacent tissue samples (see Figure 3) lends itself to the argument that DNA methylation plays a causal role in tumorigenesis. Selective TPEF inactivation by aberrant DNA methylation processes is not understood, but these data suggest that genes important for growth control can be identified in human cancers and point to the use of hypermethylated tumor suppressor genes as markers of tumorigenesis. (Dr. Weisenberger is a Postdoctoral Fellow in the Urologic Research Laboratory, Department of Biochemistry and Molecular Biology; Dr. Jones is the Director and H. Leslie & Elaine S. Hoffman Cancer Research Chair; and Dr. Liang is an Assistant Professor in the Urologic Research Laboratory, Department of Biochemistry and Molecular Biology, Norris Comprehensive Cancer Center and Hospital, Keck School of Medicine at the University of Southern California in Los Angeles.)
| Figure 2. Methylation Levels of TPEF in Tumor Cell Lines and Primary Tumors |
 |
References
1. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999;21:163-167.
2. Jones PA. DNA methylation errors and cancer. Cancer Res 1996;56:2463-2467.
3. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987;196:261-282.
4. Laird PW, Jaenisch R. DNA methylation and cancer. Hum Mol Genet 1994;3:1487-1495.
5. Robertson KD, Jones PA. DNA methylation: Past, present and future directions. Carcinogenesis 2000;21:
461-467.
6. Panning B, Jaenisch R. RNA and the epigenetic regulation of X chromosome inactivation. Cell 1998;93:
305-308.
7. Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature 1993;366:362-365.
8. Jones PA. The DNA methylation paradox. Trends Genet 1999;15:34-37.
9. Gonzalez-Zulueta M, Bender CM, Yang AS, et al. Methylation of the 5’ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995;55:4531-4535.
10. Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A 1994;91:
9700-9704.
11. Pfeifer GP, Steigerwald SD, Mueller PR, et al. Genomic sequencing and methylation analysis by ligation mediated PCR. Science 1989;246:810-813.
12. Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis.
J Mol Biol 1975;98:503-517.
13. Toyota M, Ho C, Ahuja N, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 1999;59:2307-2312.
14. Gonzalgo ML, Liang G, Spruck CH, et al. Identification and characterization of differentially methylated regions of genomic by methylation-sensitive arbitrarily primed PCR. Cancer Res 1997;57:594-599.
15. Liang G, Salem CE, Yu MC, et al. DNA methylation differences associated with tumor tissues identified by genome scanning analysis. Genomics 1998;53:260-268.
16. Liang G, Robertson KD, Talmadge C, et al. The gene for a novel transmembrane protein containing epidermal growth factor and follistatin domains is frequently hypermethylated in human tumor cells. Cancer Res 2000;60:4907-4912.
17. Eib DW, Martens GJ. A novel transmembrane protein with epidermal growth factor and follistatin domains expressed in the hypothalamo-hypophysial axis of Xenopus laevis. J Neurochem 1996;67:1047-1055.
18. Patel K. Follistatin. Int J Biochem Cell Biol 1998;30:
1087-1093.
19. Engler DA, Montelione GT, Niyogi SK. Human epidermal growth factor. Distinct roles of tyrosine 37 and arginine 41 in receptor binding as determined by site-directed mutagenesis and nuclear magnetic resonance spectrocopy. FEBS Lett 1990;271:47-50.
20. Gonzalgo ML, Jones PA. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res 1997;25:2529-2531.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.