Polyunsaturated Fatty Acids and Skin Cancer: Part II
Polyunsaturated Fatty Acids and Skin Cancer: Part II
By Anthony P. Albino, PhD and Leonard A. Cohen, PhD
To date, three fundamental observations have emerged that clearly support the thesis that n-3 PUFAs have clinical potential as an adjuvant cancer therapeutic modality: 1) n-3 PUFAs can be directly
cytotoxic to tumor cells independent of other agents;1,2 2) n-3 PUFAs can have synergistic or additive antitumor effects when used in combination with conventional chemotherapeutic agents;2-4 and 3) n-3 PUFAs can reverse intrinsic tumor cell drug resistance to several chemotherapeutic drugs (e.g., vincristine, doxorubicin, and cis-platinum).2,5
Mechanisms of Action
Developing a mechanistic basis for these multifactorial antitumor roles has been the focus of a large body of research.2,6 These studies have shown that n-3 PUFAs can exert their effects by a number of basic mechanisms. They can directly alter the composition and, thus, the basic structure of the cell’s bilipid plasma membrane, allowing facilitated entry of chemotherapeutic drugs (e.g., adriamycin, doxorubicin, and vincristine).5-7 n-3 PUFAs can increase the generation of damaging reactive oxygen species (ROS) via membrane lipid peroxidation by drugs like doxorubicin. They may alter gene expression or change the composition and bioactivity of downstream metabolites of PUFAs (i.e., the eicosanoids). These types of alterations can, in turn, induce tumor cell cytostasis or cytotoxicity by excessive DNA damage, induction of apoptosis, suppression of tumor cell-
stromal matrix association, inhibition of tumor-driven angiogenesis, and alterations in key cell cycle regulatory proteins.
Antitumor Potential of PUFAs
Cell cycle deregulation, in particular, is a hallmark of transformed cells. How PUFAs affect the cell cycle is obscure. In melanoma, as in other cell types, normal transition from one phase of the cell cycle to the next is regulated at checkpoints, which are, in part, governed by cyclin-dependent kinases (CDKs) that are assembled with partner cyclins.8 Active CDK-cyclin complexes are further regulated by phosphorylation-dephosphorylation events that are mediated through CDK-activating kinases and phosphatases. CDKs also can be inactivated through physical binding with CDK inhibitory proteins (CKIs).8 D- and E-type cyclins are important in cell cycle transition from G1 to S.9 The cyclin D-CDK complex is believed to function by specific phosphorylation of the RB product.9 Hypophosphorylated pRB binds members of the E2F transcription factor family, leading to inactivation of E2F function and reduced expression of genes critical for S-phase events (e.g., dihydrofolate reductase, DNA polymerase a, and cyclins).10 Hyperphosphorylation of pRB disables its E2F-binding, thereby influencing the passage of cells from G1 into S and cellular proliferation.
Thus, pRB is the master switch regulating cell cycle progression and its continuing phosphorylation parallels cell transit through G1 and S.10 The initial phosphorylation carried out by the Cdk4(6)/cyclin D complex is required for cell passage through the G1 restriction point.10 Although many tumor types have disruptions in the pRB pathway, virtually 100% of invasive and metastatic melanomas have detectable defects in one or more of the important regulators of the pRB regulatory circuit (i.e., the cyclin-dependent kinase inhibitor 2A [CDKN2A] or p16INK4a gene; the CDKN2B or p15INK4b gene; and the D-type cyclins and their functional partners Cdk4 and Cdk6).11,12 The most common defect in melanoma cells is the loss or mutation of the p16/CDKN2A gene; although there is only minimal dysfunction of pRB itself, with most melanoma specimens and established cell lines expressing normal RB protein.12,13 Defects in the pRB circuit appear to play a major contributory role in inducing uncoordinated tumor cell proliferation, a fundamental biological trait that differentiates the melanoma cell from the usually non-proliferative benign melanocyte.
Presently, little is known about the effects of fatty acids on cell cycle control, and we only recently have begun to elucidate the mechanism by which docosahexaenoic acid ([DHA] found in high levels in cold water fish such as mackerel, menhaden, salmon, and tuna) inhibits the growth of human melanoma cells.14 The molecular events by which DHA suppresses the growth of melanoma cells was examined in detail in two melanoma cell lines: one refractory (SK-Mel-29) and the other sensitive (SK- Mel-110) to the inhibitory effects of DHA. Exponentially growing melanoma cell lines were exposed in vitro to DHA. Then the cell lines were assessed for inhibition of cell growth; expression of cyclins and cyclin-dependent kinase inhibitors in individual cells by flow cytometry and immunocytochemistry, using specific monoclonal antibodies to cyclin D1, cyclin E, p21WAF1/CIP1, or p27KIP1; and expression of total pRBT independent of phosphorylation state and hypo-phosphorylated pRBP- in fixed cells by flow cytometry and immunocytochemistry, using specific monoclonal antibodies to pRBT or pRBP-, respectively.
Upon treatment with increasing concentrations of DHA, cell growth in seven of 12 melanoma cell lines was inhibited, whereas cell growth was minimally affected in the other five cell lines. Two melanoma cell lines were examined in detail, one that was resistant (SK-Mel-29) and one that was sensitive (SK-Mel-110) to the inhibitory activity of DHA. SK-Mel-29 cells were unaffected by treatment with up to 2 mcg/mL DHA. No appreciable change was observed in cell growth, cell cycle distribution, the status of pRB phosphorylation, cyclin D1 expression, or the levels of the CKIs, p21 and p27.
In contrast, SK-Mel-110 cell growth was inhibited by DHA with the cells accumulating either in G1 or S phase (0% in SK-Mel-29 vs 41.2% in SK-Mel-110). Moreover, considerable death occurred by apoptosis. In addition, DHA treatment resulted in increasing numbers of SK-Mel-110 cells expressing hypophosphorylated pRB (from 12% to > 40%), whereas the levels of cyclin D1 and p21 changed little.
Potential Clinical Applications
This study showing that DHA inhibits the growth of cultured metastatic melanoma cells provides information as to potential mechanistic interactions of fatty acids with specific components of the cell cycle machinery. The finding that DHA-induced growth inhibition correlates with a quantitative increase in hypophosphorylated pRB suggests a cross-talk mechanism between fatty acid metabolism and the pRB pathway. More importantly, these data suggest that reactivation of pRb by n-3 PUFAs may prevent the abnormal proliferation of melanoma cells. Determining the mechanism by which n-3 PUFAs can inhibit melanoma growth will be an important first step in the rational use of these PUFAs as antitumor agents in combination with conventional chemotherapeutic drugs.
For example, one of the most potent single-agent drugs for the treatment of melanoma is interferon-a (IFN-a). The interferons are a family of naturally occurring, small proteins with molecular weights of approximately l5,000-21,000 Da. They are produced and secreted by virtually all eukaryotic cells in response to viral infections or to various biologic and synthetic inducers.15 Three major classes of interferons have been identified: alpha, beta, and gamma.
Interferons induce their cellular activities by binding to specific membrane receptors on cell surfaces. IFN-a-2b, like the naturally occurring alpha or leukocyte interferons, demonstrates potent antiproliferative and anti-viral properties. IFN-a has shown both immunomodulatory and antiproliferative effects in metastatic mela-noma. IFN-a-2b recently has been approved by the FDA as the first effective adjuvant therapy for the treatment of the "high risk for recurrence" melanoma patient.16 The antitumor activity of IFN-a can be affected by direct and indirect mechanisms. For example, IFN can induce cytostatic or cytotoxic effects by increasing the length of the cell cycle, by affecting the expression levels of various critical oncogenes (i.e., c-myc, c-fos, or c-H-ras genes), by inhibiting the induction of enzymes critical for cell survival, or by impacting the antitumor actions of host cytotoxic T-cells.15,17,18 Each of these mechanisms would be capable of augmenting or complementing the several types of antitumor actions of n-3 PUFAs discussed above.
Thus, in theory, a combination therapy using both IFN-a and n-3 PUFA supplementation should provide a more potent antitumor effect than either agent alone. We have preliminary in vitro data showing that treatment of melanoma cells with DHA and IFN-a has synergistic inhibitory effects on their growth and motility.
Ratio of n-3 to n-6 PUFA
An important aspect of any dietary trial using PUFAs is to control the n-3:n-6 ratio. The clinical objective of n-3 PUFA supplementation is to increase the overall n-3:
n-6 PUFA ratio and to decrease the production of arachidonic acid and the subsequent formation of specific eicosanoid metabolites (which include the prosta-glandins, leukotrienes, and thromboxanes).19 PUFAs are converted to metabolites via a series of enzymatic reactions that add more double bonds to the molecules (mediated by desaturase enzymes) and elongations that extend the length of the carbon chain (mediated by elongase enzymes).20 (See Figure.) These reactions ultimately convert dietary n-6 linoleic acid to arachidonic acid (20:4n-6) and n-3 alpha linolenic acid to eicosapentaenoic acid (EPA) (20:5n-3). The subsequent metabolism of these PUFAs produces a different spectrum of eicosanoids because of the competition that exists between the n-3 and n-6 PUFAs for the D4 and D6 desaturases (n-3 PUFA having greater affinities for the enzyme active sites). Thus, increasing the dietary intake of n-3 PUFAs (e.g., alpha linolenic acid, EPA, or DHA), while simultaneously increasing the n-3:n-6 ratio, reduces the desaturation of linoleic acid and, therefore, the production of arachidonic acid.21 Simply increasing n-3 levels or decreasing n-6 levels without changing the overall n-3:n-6 ratio has been shown not to reduce the levels of arachidonic acid or its metabolites in human tissues.22
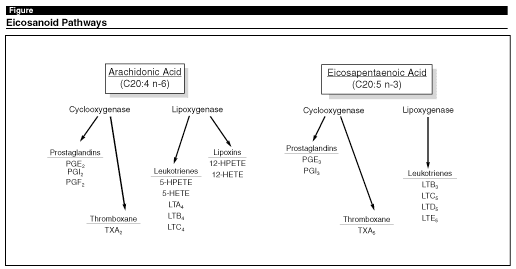 |
Altering the spectrum of eicosanoids is important because of their differing physiologic effects on tumor cells.23 In a study designed to examine the effect of PUFA on susceptibility to lung metastases in mice, Abbott and associates used experimental diets in which the predominant lipids were either n-3 PUFAs or n-6 PUFAs.24 The n-3 (fish oil) diet was found to be protective, whereas the n-6 (corn oil) diet was not. The superiority of the fish oil diet was attributed to its higher n3:
n-6 ratio and a concomitant decrease in arachidonic acid formation. Other studies also stress the importance of overall n-3:n-6 ratio in the diet rather than the absolute amount of n-3 PUFAs as a primary determinant of eicosanoid biosynthesis.25 In rats, it was found that the suppression of eicosanoid biosynthesis from arachidonic acid by n-3 PUFAs showed no dose response if the
n-3:n-6 ratio was left unchanged.25 However, a shift in the ratio from 0.3 to 0.6 was the determining factor in inhibiting the production of eicosanoids. A study designed to examine the interactions between fish and vegetable oils in affecting rat leukocyte phospholipid PUFAs and leukotriene production showed that EPA-rich dietary fish oil supplements achieved a greater reduction in leukotriene B-4 synthesis when the linoleic acid content of the total fat intake approached that of olive oil rather than safflower or corn oil.26
While there are few published data concerning the impact on oncogenesis of altering the n-3:n-6 ratio in humans, epidemiological observations support the supposition that a significant reduction in breast cancer risk occurs when the n-3:n-6 ratio is raised.27 Lands and colleagues have observed an overall inverse relationship between cancer and dietary n-3:n-6 ratio.28 In Japan, a downward shift in the estimated dietary n-3:n-6 ratio from a mean value of 0.34 in 1960 to 0.26 in 1985 was observed, with the concomitant increase in risk of breast and colon cancers. In 1985, the corresponding value for the United States was only 0.12. Rose has recently reviewed a large number of studies that used animal models to investigate the relationship of PUFAs and mammary and prostate cancers.29 He concluded that n-6 PUFAs stimulate mammary carcinogenesis and tumor growth and metastasis, whereas n-3 PUFAs exhibit inhibitory effects. Studies of prostate cancers are less advanced, but the available data suggest a similar stimulatory role for n-6 PUFAs and an inhibitory role for n-3 PUFAs. Thus, based on accumulated data, Rose makes a cogent argument for clinical intervention trials designed to reduce total fat intake and increase the n-3:n-6 PUFA ratio in groups at high risk of developing cancer, and in cancer patients in remission, with the objective of preventing disease recurrence.
Conclusion
The observation that the capacity of PUFAs to promote or suppress tumor development and progression depends more on the n-6:n-3 ratio than on the total amounts of these PUFAs in the diet indicates that n-6 and n-3 PUFAs have different, but overlapping, physiologic functions. Altering the balance of these fatty acids can affect mechanisms that control critical biological characteristics of tumor progression, such as tumor cell proliferation and metastatic potential. Manipulating the quantity and composition of dietary fatty acids may prevent and/or retard both localized growth and the development of distant metastases of a wide range of common cancers, including non-melanoma and melanoma skin cancers.
We further hypothesize that the addition of specific
n-3 PUFAs to the diets of patients with advanced cancers, in conjunction with conventional therapeutic modalities, can provide a more efficacious therapeutic treatment than standard therapy alone. Controlling dietary n-3 PUFAs in patients at high risk for tumor recurrence and metastasis represents a conceptually new approach to improve clinical outcomes in patients with advanced disease. In addition, it affords patients a potential psychological edge in being able to control and participate in their own treatment using familiar substances that appear naturally in the food supply. (Dr. Albino is the Director of Research and Dr. Cohen is the Section Head, Nutrition and Endocrinology, The American Health Foundation, Valhalla, NY.)
References
1. Burns CP, Spector AA. Effects of lipids on cancer therapy. Nutr Rev 1990;48:233-240.
2. Plumb JA, Luo W, Kerr DJ. Effect of polyunsaturated fatty acids on the drug sensitivity of human tumour cell lines resistant to either cisplatin or doxorubicin.
Br J Cancer 1993;67:728-733.
3. Germain E, Chajes V, Cognault S, et al. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast cancer cell line MDA-MB-231: Relationship to lipid peroxidation. Int J Cancer 1998;75:578-583.
4. Needs GT, Jones DL, Hughes LE. Effect of polyunsaturated fatty acids on doxorubicin sensitivity in MCF-7 cells. Br J Cancer 1991;78:755.
5. Das UN, Madhavi N, Sravan Kumar G, et al. Can tumour cell drug resistance be reversed by essential fatty acids and their metabolites? Prostaglandins Leukot Essent Fatty Acids 1998;58:39-54.
6. Burns CP, North JA. Adriamycin transport and sensitivity in fatty acid-modified leukemia cells. Biochim Biophys Acta 1986;888:10-17.
7. Burns CP, North JA, Petersen ES, et al. Subcellular distribution of doxorubicin: Comparison of fatty acid-modified and unmodified cells. Proc Soc Exp Biol Med 1988;188:455-460.
8. Sherr CJ. Cancer cell cycles. Science 1996;274:
1672-1677.
9. Sherr CJ, Kato J, Quelle DE, et al. D-type cyclins and their cyclin-dependent kinases: G1 phase integrators of the mitogenic response. Cold Spring Harb Symp Quant Biol 1994;59:11-19.
10. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323-330.
11. Sellers WR, Kaelin WG Jr. Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol 1997;15:3301-3312.
12. Walker GJ, Flores JF, Glendening JM, et al. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer 1998;
22:157-163.
13. Maelandsmo GM, Florenes VA, Hovig E, et al. Involvement of the pRb/p16/cdk4/cyclin D1 pathway in the tumorigenesis of sporadic malignant melanomas. Br J Cancer 1996;73:909-916.
14. Albino AP, Juan G, Traganos F, et al. Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid: Association with decreased pRB phosphorylation. Cancer Res 2000;60:4139-4145.
15. Hanley JP, Haydon GH. The biology of interferon-alpha and the clinical significance of anti-interferon antibodies. Leuk Lymphoma 1998;29:257-268.
16. Kirkwood JM, Strawderman MH, Ernstoff MS, et al. Interferon alpha-2b adjuvant therapy of high risk resected cutaneous melanoma: The Eastern Cooperative Group Trial EST 1684. J Clin Oncol 1996;14:7-17.
17. Contente S, Kenyon K, Rimoldi D, et al. Expression of gene rrg is associated with reversion of NIH 3T3 transformed by LTR-c-H-ras. Science 1990;249:796-798.
18. Giacomini P, Fraioli R, Nistico P, et al. Modulation of the antigenic phenotype of early-passage human melanoma cells derived from multiple autologous metastases by recombinant human leukocyte, fibroblast and immune interferon. Intl J Cancer 1990;46:
539-545.
19. Mantzioris E, James MJ, Gibson RA, et al. Differences exist in the relationships between dietary linoleic acid and a-linolenic acids and their respective long-chain metabolites. Am J Clin Nutr 1995;61:320-324.
20. De Gomez Dumm INT, Brenner RR. Oxidative desaturation of alpha-linolenic, linoleic, and stearic acids by human liver microsomes. Lipids 1975;10:315-317.
21. Cleland LG, James MJ, Neumann MA, et al. Linoelate inhibits EPA incorporation from dietary fish oil supplements in human subjects. Am J Clin Nutr 1992;55:
395-399.
22. James MJ, Gibson RA, D’Angelo M, et al. Simple relationships exist between dietary linoleate and the n-6 fatty acids of human neutrophile and plasma. Am J Clin Nutr 1993;58:497-500.
23. Kramer HJ, Stevens J, Grimminger F, et al. Fish oil fatty acids and human platelets: Dose-dependent decrease in dienoic and increase in trienoic thromboxane generation. Biochem Pharmacol 1996;25:
1211-1217.
24. Abbott WG, Tezabwala B, Bennett M, et al. Melanoma lung metastases and cytolytic effector cells in mice fed antioxidant-balanced corn oil or fish oil diets. Nat Immun 1994;13:15-28.
25. Boudreau MD, Chanmugam PS, Hart SB, et al. Lack of dose response by dietary n-3 fatty acids at a constant ratio of n-3 to n-6 fatty acids in suppressing eicosanoid biosynthesis from arachidonic acid. Am J Clin Nutr 1991;54:111-117.
26. James MJ, Cleland LG, Gibson RA, et al. Interaction between fish and vegetable oils in relation to rat leukocyte leukotriene production. J Nutr 1991;121:631-637.
27. Kaizer L, Boyd NF, Kriukov V, et al. Fish consumption and breast cancer: An ecological study. Nutr Cancer 1989;12:61-68.
28. Lands WE, Hamazaki T, Yamazaki K, et al. Changing dietary patterns. Am J Clin Nutr 1990;51:991-993.
29. Rose DP. Dietary fatty acids and prevention of hormone-responsive cancer. Proc Soc Exp Biol Med 1997;
216:224-233.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.