Loss of TAP and LMP Expression in Renal Cell Carcinomas: A Mechanism of Tumor Escape
Loss of TAP and LMP Expression in Renal Cell Carcinomas: A Mechanism of Tumor Escape
By Nilanjan Ghosh and Kenneth L. Wright, PhD
The major histocompatibility complex class i (MHC-I) molecules present endogenous antigenic peptides to the cytotoxic CD8+ T cell population. This interaction between the antigen-presenting cells and the cytotoxic T cells is required for T cell-mediated killing of the infected or transformed cells. Endogenous proteins are degraded to peptide fragments in the cytosol, and peptides with a preferred length of 7-13 amino acids are transported into the endoplasmic reticulum (ER). In the ER or in the cis-Golgi, a stable trimolecular complex is formed, composed of a peptide, a newly synthesized MHC-I molecule, and b2-microglobulin (b2m). This complex then is transported to the cell surface. A CD8+ T cell, bearing a T cell receptor (TCR) that specifically recognizes a given MHC-I allele with a uniquely bound peptide, will be stimulated to proliferate and become activated by the interaction. The activated T cells then kill the antigen-presenting cell through the action of perforin and granzymes, thus eliminating the aberrant cells from the body.
TAP1, TAP2, LMP2, and LMP7: Functional Role in the MHC-I Antigen-Presenting Pathway
The proteosome is a multi-catalytic, multi-subunit complex previously demonstrated to be part of the ubiquitin-dependent protein degradation pathway present in all cells. Two proteosome subunits, low molecular mass polypeptide 2 and 7 (LMP2 and LMP7) and a third molecule, MC14 (or LMP9) associate with the 20 s proteosome complex, following induction by the cytokine interferon gamma (IFN-g).1,2 Incorporation of LMP2, LMP7, and MC14 into the complex displaces constitutive subunits and modulates the types of peptides generated.3,4 These proteosomes have been called immunoproteosomes because the specificity of the proteosome complex is altered to enhance the production of peptides cleaved after hydrophobic and basic residues. These peptides often are preferred by MHC-I molecules. Mice deficient in either LMP2 or LMP7 show susceptibility to specific infections due to failure to generate important immunodominant epitopes. Thus, association of LMP2 and LMP7 with the proteosome is important for effective and complete antigen processing and presentation.
The transporters associated with antigen processing (TAP1 and TAP2) are heterodimers that reside in the ER and cis-Golgi membranes.5-7 TAP molecules transport antigenic peptides in an ATP-dependent manner into the ER lumen for binding by the MHC-I heavy chain. TAP physically associates with the MHC-I/b2M dimer through a bridging molecule, tapasin, during peptide loading in the ER.8 Calnexin and calrericulin are present as molecular chaperones during function of the trimolecular complex. A schematic of MHC-I antigen-presenting pathway is depicted in Figure 1.
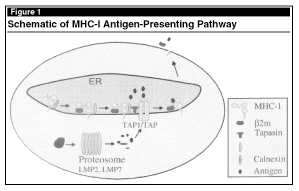 |
Studies on TAP-deficient or mutant cell lines have been invaluable for understanding the importance of TAP in the MHC-I antigen-presenting pathway. Loss of TAP can lead to the absence of MHC-I expression on the cell surface, despite the presence of MHC-I mRNA or a selective inability to present endogenous antigens.9,10 TAP 1-deficient mice lack MHC-I expression on the cell surface and have severe deficiency in CD8+ T cells.11
Tumor Escape
Recognition of the peptide-MHC-1 complex by effector T cells leads to a sequence of events that results in lysis of the transformed cell. Thus, T cells can help regulate tumor progression in an immunocompetent host. However, tumor cells have developed mechanisms to escape recognition from the patrolling cytotoxic T cells. This escape from immune surveillance may lead to uninhibited proliferation of the transformed cells, de-spite the presence of tumor-reactive T cells. One direct mechanism of tumor escape is to prevent expression of MHC-I molecules with tumor-associated peptides on the cell surface. This could be accomplished by inhibiting the expression or directly mutating the MHC-I, TAP, or LMP molecules. In addition, since each of these genes is up-regulated by interferon-gamma (IFN-g), inhibiting the IFN-g signaling pathway would significantly inhibit antigen presentation. This ultimately may render the tumor cells resistant to lysis by MHC-I antigen-restricted cytotoxic T cells and allow tumor progression.
Transcriptional Regulation of TAP and LMP Genes
All of the TAP1, TAP2, LMP2, and LMP7 genes are adjacent to each other and located within the MHC locus on human chromosome 6. The TAP1 and LMP2 genes are transcribed from a shared bidirectional promoter. A minimal 593 bp region separating the ATG translation initiation codons of both these genes is sufficient for concurrent expression in both directions.12 The absence of TATA boxes is well correlated with the presence of multiple start sites for both these genes. The transcriptional regulation of these genes is mediated by the cis-acting elements in the promoter region proximal to the TAP1 gene. They include an interferon response element and a gamma-activated sequence (IRF-E/GAS) composite site that is bound both by the interferon response factor-1 (IRF-1) and the signal transducer and activator of transcription 1 alpha (STAT1a). Both IRF-1 and STAT1 activity are stimulated by IFN-g treatment. Recently, it also has been demonstrated that unphosphorylated STAT1 and IRF-1 can form a complex and bind to the promoter region to mediate constitutive LMP2 tran-scription.13 The TAP1/LMP2 promoter also utilizes an SP1 binding site for constitutive activity and a NFkB site, which can respond to multiple stimuli, including TNF-a.
INF-g Signaling Pathway
The pathway of IFN-g signaling to gene activation has been elegantly deciphered in the last several years (see Figure 2). IFN-g, a cytokine secreted by T cells and natural killer cells, has antiproliferative effects and induces MHC-I, TAP1, TAP2, LMP2, and LMP7 genes. IFN-g treatment of certain renal carcinoma cell (RCC) lines leads to induction of TAP1 and LMP2 genes via the IRF-E/GAS DNA element proximal to the TAP1 promoter, and mutation of this element abolishes the inducibility. This TAP and LMP induction, which is independent of new protein synthesis, is followed by MHC-I expression.14 This may be responsible for enhanced recognition of RCC by the immune system.
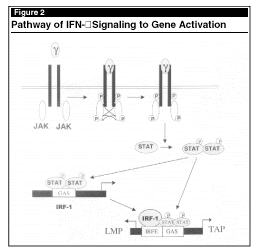 |
The binding of IFN-g to its receptor leads to activation and phosphorylation of the receptor-associated Janus kinases (Jak1 and Jak2). Receptor tyrosine phosphorylation generates a binding site for STAT1a, which is subsequently phosphorylated by Jak1 and Jak2 to form active STAT1a homodimers. Activated STAT1a translocates to the nucleus where it binds to the DNA at GAS elements in the promoters of many IFN-g-induced genes including the IRF-1 gene promoter. IRF-1 expression is induced subsequently.15 The IRF-1 protein binds to IRF-E DNA elements in additional IFN-g-induced genes. The TAP1/LMP2 promoter utilizes both direct induction by STAT1a and secondary induction by IRF-1 at its composite IRF-E/GAS DNA element. Induction of TAP and LMP by IFN-g could assist in overcoming the expression of unstable MHC-I molecules found in multiple tumor types, including several RCC lines.9,10,14 Thus, restoring the antigen-presenting machinery by inducing TAP and LMP may improve the cytotoxic T cell-mediated, antigen-specific antitumor effect in patients bearing carcinomas with TAP and LMP deficiency.
Inability of IFN-g to Induce TAP1 and LMP2 in Renal Carcinoma Cell Line Caki-2
Recently it has been shown that RCC cell lines Caki-1 and Caki-2 show low constitutive levels of TAP1 and LMP2.10 On IFN-g treatment, TAP1 and LMP2 were induced only in Caki-1 cells, but not in Caki-2. IFN-g resulted in a two- to three-fold induction of TAP1 and LMP2 genes in Caki-1 cells, but had no effect in Caki-2 cells.16 Consistent with previous results, IFN-g was unable to induce TAP and LMP expression in either cell line when the IRF-E site was mutated. Because STAT1 and IRF-1 have been shown to be involved in the transcriptional regulation of TAP1 and LMP2 genes, we investigated whether the loss of TAP1 and LMP2 expression in the RCC cell line, Caki-2, was attributable to deficiencies of these factors.16
IRF-1 Is Not Bound to the IRF-E Site in Caki-2 Cells
In vivo genomic footprinting is an excellent technique for examining DNA-protein interactions within the cell. Briefly, live cells are treated with dimethyl sulfate (DMS), which permeates through the membranes into the nucleus and methylates the guanine residues in DNA. However, if a protein is bound to DNA, the guanine residue can be protected from methylation. Then the DNA is isolated from the cells and treated with piperidine, which cleaves at the methylated guanine residues, leading to formation of a DNA ladder. The gene of interest is amplified by PCR and analyzed on a sequencing gel. The results are compared to deproteinated DNA from the same cells, which was isolated prior to treatment by DMS. By using this technique we determined that the Caki-1 cells showed a strong protection of guanine residues upon IFN-g induction at the IRF-E site in the bidirectional promoter region, indicating that this site was occupied in vivo. However, in Caki-2 cells this site was not occupied upon IFN-g induction.
To determine the identity of the proteins bound to the promoter region in these cells, we looked at in vitro DNA protein-binding ability by electrophoretic mobility shift assays. A radio-labeled oligonucleotide containing the IRF-E element from the TAP1/LMP2 promoter was incubated with nuclear extracts from Caki-1 and Caki-2 cells. After five hours of IFN-g treatment, protein bound to the IRF-E site was easily detected in the Caki-1 cells. However IFN-g treatment failed to induce IRF-1 DNA binding activity in Caki-2 cells.
Mechanism of IRF-1 Down-Regulation in Caki-2 Cells
Activation and phosphorylation of STAT1 is required for IRF-1 induction by IFN-g. Since activated STATs form homodimers and bind to DNA, this phenomenon can be used as a tool for determining the presence of activated STAT proteins in the cell. No activated STAT1 protein DNA binding activity was detectable in Caki-2 cells as opposed to Caki-1 cells. Activation of STAT1 also can be measured directly by using antibodies that specifically recognize the phosphorylated form of STAT1. Upon IFN-g treatment, phosphorylated STAT1 is induced in Caki-1 cells, but not in Caki-2 cells. However, constitutive levels of unphosphorylated STAT1 are detectable in both Caki-1 and Caki-2 cells. This indicates that the defect in Caki-2 cells lies not in the expression of STAT1, but rather in the activation of STAT1. Tyrosine phosphorylation of Jak1 and Jak2 induces STAT1 activation via the IFN-g signaling pathway. Induction of Jak1 and Jak2 phosphorylation is seen in Caki-1 cells, but not in Caki-2 cells. However, constitutive levels of Jak1 and Jak2 proteins are detectable in both cell lines. There are two possibilities to explain this phenomenon. Either there is a mutation in the Jak1 and/or Jak2 proteins which prevents them from being activated, or there is a mutation in the IFN-g receptor rendering the Caki-2 cell line unresponsive to IFN-g. Overexpression of Jak1 and/or Jak2 was not able to re-establish inducibility of the Caki-2 cell line to IFN-g. Thus, the defect in this cell line is not due to abnormal Jak proteins. Although IFN-g receptor is expressed in the Caki-2 cell line, it appears that mutations in the receptor hamper the IFN-g signaling pathway in these cells.
Role of IFN-g as an Immunotherapeutic Agent in Renal Cell Carcinoma
Patients with defective IFN-g signaling are susceptible to severe disseminated infections.17,18 Importantly, IFN-g sensitivity is required for the enhancement of tumor immunogenicity and elicitation of an effective tumor-specific immune response. A large proportion of tumors show IFN-g unresponsiveness,19 and these tumors may be able to circumvent detection by the host immune system. RCC, an epithelial cell tumor, is the most common malignancy of the kidney. The incidence has been slowly rising and it is estimated that in the United States, 31,200 people will be diagnosed with kidney cancer in the year 2000, and 11,900 will die of the disease.20 At diagnosis, 55% of patients have either metastatic disease or locally advanced tumors with lymph node and/or local organ involvement.21 Patients with metastatic disease have a three- to five-year survival rate of less than 5%.22
The rationale for using immunotherapy for treating advanced and metastatic RCC came from the observation that many of these tumors demonstrated infiltration by lymphocytes and macrophages.23 In 25% of patients, these tumor-infiltrating lymphocytes were able to lyse autologous tumor cells in a MHC-restricted manner and/or produce IFN-g in response to autologous tumor but not allogenic RCC.24 A group of RCC is relatively responsive to cytokines with growth inhibitory and immunomodulating properties, such as IFN-a and IFN-g. Low-dose IFN-g treatment was effective in maintaining long-term complete remissions in approximately 15% of RCC patients with limited disease.25-27 The up-regulation of TAP and LMP molecules by IFN-g leading to an effective MHC-I processing machinery may be one mechanism by which low-dose IFN-g therapy has its effect.
This hypothesis is further supported by the demonstration that transfer of TAP1 gene in a RCC cell line enhanced its immunogenicity.28 However, the observations that some RCCs do not up-regulate TAP and LMP in response to IFN-g and our findings that the IFN-g signaling pathway can be defective in these tumors, indicate that examining the IFN-g response in RCC patient samples could be informative to estimate the likely success of low-dose IFN-g.
Conclusion
Studies of TAP1 and LMP2 transcriptional regulation in tumor cells have provided us essential information for designing genetic and immunotherapeutic strategies to enhance MHC-I antigen presentation to combat cancer. One such strategy would be to screen tumor samples obtained during biopsy or surgery in RCC patients to design specific therapeutic measures. A simple schematic is described in Figure 3. (Mr. Ghosh is a PhD Candidate, Department of Biochemistry and Molecular Biology, Interdisciplinary Oncology Program, College of Medicine; Dr. Wright is an Assistant Professor, Interdisciplinary Oncology Program, and Member in Residence, H. Lee Moffitt Cancer Center, University of South Florida in Tampa.)
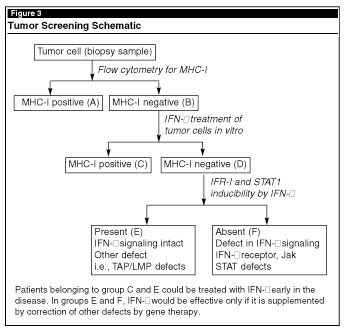 |
References
1. Kelly A, Powis SH, Glynne R, et al. Second proteasome-related gene in the human MHC class II region. Nature 1991;353:667-668.
2. Glynne R, Powis SH, Beck S, et al. A proteasome-related gene between the two ABC transporter loci in the class II region of the human MHC. Nature 1991;353: 357-360.
3. Driscoll J, Brown MG, Finley D, et al. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature 1993;365:262-264.
4. Gaczynska M, Rock KL, Goldberg AL. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 1993;365:264-267.
5. Trowsdale J, Hanson I, Mockridge I, et al. Sequences encoded in the class II region of the MHC related to the ABC’ superfamily of transporters. Nature 1990; 348:741-744.
6. Spies T, Bresnahan M, Bahram S, et al. A gene in the human major histocompatibility complex class II region controlling the class I antigen presentation pathway. Nature 1990;348:744-747.
7. Deverson EV, Gow IR, Coadwell WJ, et al. MHC class II region encoding proteins related to the multidrug resistance family of transmembrane transporters. Nature 1990;348:738-741.
8. Ortmann B, Androlewicz MJ, Cresswell P. MHC class I/beta 2-microglobulin complexes associate with TAP transporters before peptide binding. Nature 1994; 368:864-867.
9. Seliger B, Maeurer MJ, Ferrone S. TAP off—tumors on. Immunol Today 1997;18:292-299.
10. Seliger B, Hohne A, Knuth A, et al. Reduced membrane major histocompatibility complex class I density and stability in a subset of human renal cell carcinomas with low TAP and LMP expression. Clin Cancer Res 1996;2:1427-1433.
11. White LC, Wright KL, Felix NJ, et al. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8+ T cells in IRF-1-/- mice. Immunity 1996;5: 365-376.
12. Wright KL, White LC, Kelly A, et al. Coordinate regulation of the human TAP1 and LMP2 genes from a shared bidirectional promoter. J Exp Med 1995;181: 1459-1471.
13. Chatterjee-Kishore M, Wright KL, Ting JP, et al. How Stat1 mediates constitutive gene expression: A complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J 2000;19: 4111-4122.
14. Seliger B, Hammers S, Hohne A, et al. IFN-gamma-mediated coordinated transcriptional regulation of the human TAP-1 and LMP-2 genes in human renal cell carcinoma. Clin Cancer Res 1997;3:573-578.
15. Pine R, Canova A, Schindler C. Tyrosine phosphorylated p91 binds to a single element in the ISGF2/IRF-1 promoter to mediate induction by IFN alpha and IFN gamma, and is likely to autoregulate the p91 gene. EMBO J 1994;13:158-167.
16. Dovhey SE, Ghosh NS, Wright KL. Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res 2000;60:5789-5796.
17. Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest 1998;101:2364-2369.
18. Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996; 335:1941-1949.
19. Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 1998;95:7556-7561.
20. American Cancer Society. Available at: http://www3.cancer.org/cancerinfo/load_cont.asp?ct=22&doc=30&Language=English. Accessed on Feb. 1, 2001.
21. Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology (Huntingt) 1999;13:801-810.
22. Golimbu M, Joshi P, Sperber A, et al. Renal cell carcinoma: Survival and prognostic factors. Urology 1986; 27:291-301.
23. Finke JH, Tubbs R, Connelly B, et al. Tumor-infiltrating lymphocytes in patients with renal-cell carcinoma. Ann N Y Acad Sci 1988;532:387-394.
24. Finke JH, Rayman P, Hart L, et al. Characterization of tumor-infiltrating lymphocyte subsets from human renal cell carcinoma: Specific reactivity defined by cytotoxicity, interferon-gamma secretion, and proliferation. J Immunother Emphasis Tumor Immunol 1994; 15:91-104.
25. Garnick MB, Reich SD, Maxwell B, et al. Phase I/II study of recombinant interferon gamma in advanced renal cell carcinoma. J Urol 1988;139:251-255.
26. Aulitzky W, Gastl G, Aulitzky WE, et al. Successful treatment of metastatic renal cell carcinoma with a biologically active dose of recombinant interferon-gamma. J Clin Oncol 1989;7:1875-1884.
27. Bukowski RM, Novick AC. Clinical practice guidelines: Renal cell carcinoma. Cleve Clin J Med 1997;64:413.
28. Kallfelz M, Jung D, Hilmes C, et al. Induction of immunogenicity of a human renal cell carcinoma cell line by TAP1-gene transfer. Intl J Cancer 1999;81: 125-133.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.