Stroke in Primary Care Patients, Part I: Risk Factors, Clinical Presentation, and Pathophysiology
Stroke in Primary Care Patients
Part I: Risk Factors, Clinical Presentation, and Pathophysiology
Authors: Alan Z. Segal, MD, Assistant Professor, Department of Neurology, Weill Medical College, Cornell University, Ithaca, NY; and Giancarlo DePasquale, MD, Research Fellow, Cornell Stroke Program, Weill Medical College, Cornell University, Ithaca, NY.
Editor’s Note—The acute onset of a focal neurologic deficit or disturbance in level of arousal should raise suspicion of ischemic stroke. Stroke is the clinical manifestation of the acute infarction of neurons secondary to ischemia. Ischemic stroke may present in a heterogenous fashion presenting a challenge to both diagnosis and treatment.
Epidemiology
Stroke is the third most common cause of death in the United States, after heart disease and all cancers, and is the most common cause of severe disability, accounting for a large proportion of health care resource usage.1 Its effect on individual patients, their families, and society is immense. About 200 previously healthy people per 100,000 population will suffer a stroke every year. Data compiled by the Prevention Patient Outcomes Research Team, indicate that annually there are as many as 550,000 hospitalizations and 150,000 deaths attributable to stroke in the United States.2 Men and women are affected in roughly equal numbers with a mean age of 72 years. Despite the uncertainty over whether stroke incidence is rising, falling, or remaining the same, the absolute number of patients is likely to increase, as incidence increases with age and the overall population becomes older.
The economic burden of stroke can be defined in terms of the direct costs of providing medical care to patients and the indirect costs associated with loss of productivity. The most recent prevalence-based study of the cost of stroke in the United States estimated this cost to be $30 billion in 1993: $17 billion in direct medical costs and $13 billion in indirect costs associated with lost earnings.3 The lifetime cost per person suffering a stroke is approximately $100,000.4
Risk Factors
Several conditions and lifestyle factors have been identified as risk factors for stroke. Modifiable risk factors include hypertension, diabetes mellitus, tobacco use, hypercholesterolemia, and alcohol abuse (see Table 1). Atrial fibrillation (AF) and carotid artery stenosis, well-recognized stroke risk factors, are discussed in the section on stroke etiology below. Nonmodifiable risk factors for stroke include age, sex, race, and genetic predisposition.5 Although these risk factors cannot be changed, they nonetheless serve as important identifiers of patients at risk for stroke, for whom an aggressive search for other modifiable risk factors might be particularly important.6,7
| Table 1. Stroke Risk Factors |
| Nonmodifiable |
| • Older age |
| • Male gender |
| • Race |
| • Genetic factors |
| Modifiable |
| • Hypertension |
| • Diabetes mellitus |
| • Hyperlipidemia |
| • Tobacco use |
| • Excessive alcohol consumption |
| • Physical inactivity |
Hypertension affects approximately 43 million men and women in the United States and is the most prevalent and modifiable risk factor for stroke. Hypertension is a particularly concerning risk factor in African-Americans and among the elderly. A study of men and women in Rochester, Minn, demonstrated that 55% of strokes occurred in those 75 years or older.8 Stroke risk may be substantially reduced by controlling both systolic and diastolic blood pressure.9,10 This includes reduction of isolated systolic hypertension to less than 160 mm Hg in the elderly.11
Diabetes mellitus is the most prevalent endocrinologic problem in primary care practice and is a well-established risk factor for stroke.12,13 Diabetes mellitus may increase the risk of thromboembolic stroke through multiple mechanisms. These include acceleration of large artery atherosclerosis via glycosylation-induced injury, adverse effects on both low-density lipoprotein and high-density lipoprotein cholesterol levels, and promotion of plaque formation through hyperinsulinemia. Intensive glucose control in both type 1 and type 2 diabetes has been proven to reduce microvascular complications of diabetes such as retinopathy, nephropathy, and neuropathy.14,15 In contrast, tight glucose control has not been shown to reduce macrovascular complications such as stroke. In the setting of acute stroke, hyperglycemia has been shown to produce poorer outcomes in diabetic patients. In particular, high-serum glucose in the setting of treatment with rtPA is associated with adverse outcomes. Therefore, it is prudent to promote tight glucose control both chronically and particularly in the acute setting.16
Cigarette smoking is an independent risk factor for ischemic stroke.17,18 In a meta-analysis of 32 studies, the relative risk of stroke for smokers is 1.5 (95% CI, 1.4-1.6). Stroke risk increases 2-fold among heavy smokers compared with light smokers. Passive exposure to cigarette smoke increases the progression of atherosclerosis. Further, cigarette smoking is an independent determinant of carotid artery plaque thickness, may increase blood viscosity and coagulability, enhance platelet aggregation, and elevate blood pressure.19
Hypercholesterolemia, although widely accepted as a risk factor for stroke, has never been conclusively proven to increase stroke risk. It is also conventional wisdom that lipid-lowering therapy with "statin" drugs will reduce stroke risk. Data addressing this question come exclusively from trials of primary and secondary prevention of coronary disease. In these studies, which are limited to individuals with coronary disease, analyses are limited to stroke as a secondary end point or as an end point determined by post-hoc analyses.20 Meta-analyses of these trials have found significant reductions in stroke risk. A 29% reduced risk of stroke and a 22% reduction in overall mortality were found, even among patients with only moderately elevated lipids.21 Stroke patients with cholesterol levels above 200 should have complete lipid profiles performed. Goal LDL of less than 130 or possibly less than 100 may be reasonable goals in patients who have had prior stroke/transient ischemic attack (TIA). Clinical trials of statins among patients with stroke/TIA are ongoing.
Alcohol consumption has a direct dose-dependent effect on the risk of hemorrhagic stroke.22 A J-shaped relationship between alcohol use and ischemic stroke has been proposed with a protective effect in light or moderate drinkers and an elevated stroke risk with heavy alcohol consumption.23 Alcohol may increase the risk of stroke through various mechanisms that include induction of hypertension, a hypercoagulable state, cardiac arrhythmias, and a reduction in cerebral blood flow. Light to moderate drinking may have a beneficial effect by increasing high-density lipoprotein cholesterol levels and decreasing platelet aggregation and fibrinogen levels.24
Another potentially treatable risk factor for stroke is elevated serum homocysteine, as it is associated with deficiency of dietary intake of folate, vitamin B6, and vitamin B12.25 There are few prospective data linking homocysteine levels and stroke risk, but data from the third NHANES survey and case-control studies are suggestive.26,27 Inherited forms of hyperhomocysteinemia are also associated with increased stroke risk and are discussed below.
The risks and benefits of postmenopausal hormone replacement therapy (HRT) with regard to stroke are poorly understood. There are some practitioners who would discontinue HRT after TIA or stroke. The Women’s Estrogen for Stroke Trial (WEST) showed no decrease in all-cause mortality or recurrent cerebral ischemia among women taking ERT after an initial stroke or TIA.28 Decisions to place women with or at risk for stroke/TIA on these agents, therefore, represent a complex risk/benefit calculus, factoring in the clear benefits for osteoporosis and cardiovascular risks.
Regular exercise has well-established benefits for reducing the risk of premature death and other cardiovascular disease. The beneficial effect of lowering the risk of stroke has been described predominately among whites and is more apparent for men than women and younger rather than older adults.29 A dose-response relationship between increasing amounts of physical activity and the reduction in the risk of stroke has not been shown consistently. Nevertheless, regular physical activity reduces the risk of known stroke risk factors such as coronary heart disease, hypertension, obesity, and diabetes mellitus.30
Despite the wealth of data concerning the importance of controlling stroke risk factors, management of these conditions remain challenging due to poor patient adherence as well as decreased detection and treatment by health care providers. Further reductions in the risk of stroke among patients with TIA will require enhancements in our ability to detect, modify, and treat cerebrovascular risk factors.
TIA
Approximately 300,000 TIAs occur each year in the United States.31,32 About 15% of patients experiencing stroke report a history of TIA.33 Effective prevention of subsequent stroke in patients with TIAs could, therefore, significantly reduce the overall stroke incidence. Symptoms such as transient monocular blindness, sudden weakness, or numbness may be short lived (5-10 min) or may last longer, becoming difficult to distinguish from stroke. Neurologic deficits lasting fewer than 24 hours, are classified as TIAs. Until the deficit has resolved, these patients should be viewed as having suffered a stroke. The longer the duration of symptoms, the more likely that TIA symptoms will indeed result in stroke. In the carotid territory, mean duration of TIA is about 14 minutes and, in the vertebrobasilar circulation, 8 minutes. If symptoms persist for more than 1 hour, only a minority will resolve by 24 hours.34 TIA should be considered as the harbinger of stroke and must be taken seriously. One-half of all strokes that follow TIAs occur within a year of the last TIA, making TIA an urgent medical condition requiring early evaluation and intervention. Prior data on short-term prognosis after TIA come from 2 old, relatively small population-based studies.35,36 In a more recent study based on a sample of 1707 patients with a diagnosis of TIA, the 90-day stroke risk was 10.5%, more than 50 times that expected in a cohort of similar age. One-half of the strokes occurred within 2 days of the TIA. Risk factors for recurrent cerebral ischemia included: age older than 60, diabetes, symptom duration longer than 10 minutes, and the presence of speech impairment. Short-term risks of cardiovascular events, death, and recurrent TIA were high, with a combined 25.1% risk during the 3 months following TIA.37
Diagnosis of TIA is often problematic. It may be difficult to determine whether focal neurologic symptoms are due to ischemia from impaired cerebral circulation or due to seizure or migraine. TIA may also produce small areas of infarction on diffusion-weighted MRI imaging, despite the clearance of the deficit clinically.38 In this situation, TIA may be considered to be a variant of stroke, rather than a separate syndrome. While some interventions are known to be effective after TIA, such as antiplatelet agents, anticoagulation for atrial fibrillation (AF), and endarterectomy for symptomatic carotid artery stenosis, it is not known whether more urgent therapy is justified. Treatment of TIA will be further discussed in Part II of this series.
Stroke—Clinical Presentation and Localization
Broadly, ischemic stroke may be divided into lacunar and nonlacunar (or large vessel) types. As shown in Figure 1, lacunar strokes account for 26% of all strokes. Large-vessel strokes may be caused by localized atherosclerosis (9%) or cardioembolism (19%). A large proportion of large-vessel strokes (41%) are without an explanation and may be considered cryptogenic.
| Figure 1 |
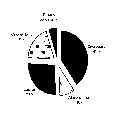 |
Lacunar strokes. Lacunar strokes are strongly associated with diabetes and chronic hypertension. Lacunar strokes are small subcortical infarcts due to single perforating artery occlusion, which may involve the vessels penetrating the brain to supply the capsule, basal ganglia, thalamus, and paramedian regions of the brain stem. Lacunar strokes may present as dense hemiparesis or sensory loss but may also occur in a subclinical, asymptomatic fashion. A large burden of such strokes may lead to multi-infarct dementia. Because nearly all of these infarcts are located within the white matter, where axon bundles travel in compact formation, small lesions can cause weakness of large portions of the body (eg, face, arm, and leg). Typical lacunar syndromes include pure motor hemiparesis (PMH), pure sensory stroke, dysarthria clumsy hand, and ataxic hemiparesis.39 Treatment for these small strokes remains controversial. Lacunar strokes have been included in nearly every major multicenter trial of intravenous thrombolysis for stroke. While no trial has examined sufficient numbers of lacunar stroke to achieve statistically significant results, these patients may achieve more rapid and complete recovery after treatment with IV rtPA. Nonetheless, because the prognosis for full or nearly full recovery is excellent without treatment, with low mortality and recurrence rates in hospital and community-based studies, lacunar infarcts do not require as aggressive management as do large-vessel strokes.40,41 Until more data are available, ASA 325 mg q.d. is probably the best therapy because of the low risk associated with this treatment. Other antiplatelet agents also might be considered for lacunar strokes. Antiplatelet therapy will be discussed in further detail in Part II (Treatment strategies for Stroke/TIA).
In some instances, lacunar strokes may be larger than commonly expected or occur on a basis other than small-vessel lipohyalinosis. Some authors have therefore suggested that there is a "fallacy to the lacunar hypothesis." These authors contend that lacunar strokes should be considered small strokes, affecting white-matter tract, not the result of a specific mechanism. Indeed, a signficiant proportion of lacunar-type strokes may occur on an embolic basis.42 Lacunar stroke has been shown to complicate cardiac catheterization or cardiac bypass, occurring on an atheroembolic basis from a diseased aorta.43
Large artery occlusive disease can be usefully divided into disorders of the anterior (internal carotid artery and its branches) and posterior (vertebral or basilar artery) circulations. Clinical presentation is the result of sudden impairment of blood flow. Unlike lacunes, these infarcts commonly involve gray matter and may be large. They carry the risk of swelling and hemorrhagic conversion. Their prognosis is much worse than that of lacunes.
In the anterior circulation, occlusion of the middle cerebral artery (MCA) is the most common site of stroke. Complete occlusion of the proximal MCA is characterized by weakness of the contralateral face, arm and leg, and hemianopia. Deviation of the eyes and head toward the side of the involved hemisphere is a distinctive and valuable sign of MCA involvement. Additional findings include aphasia if the dominant hemisphere is affected and neglect (in which the patient appears to ignore one side of his body or of his surroundings) in the nondominant hemisphere. Involvement restricted to portions of the MCA (such as distal branches only) may produce certain components of this syndrome, most specifically paralysis of the face and arm, sparing the leg. Less common than MCA occlusion, stroke in the distribution of the anterior cerebral artery (ACA), results in isolated weakness of the leg and foot. If both ACAs are affected, a generalized decrease in initiative (abulia) may occur.
Infarcts in the distribution of the posterior circulation involve the brainstem, cerebellum, thalamus, and occipital lobes. They can present with bilateral limb weakness or sensory disturbance, cranial nerve abnormalities, ataxia, nausea and vomiting, and coma. These syndromes can result from occlusion of the basilar artery or one of its branches.
Border zone or watershed infarction is the result of insufficient flow to distal territories of the major cerebral vessels. This develops most commonly in the setting of hypotension. Because the cerebral circulation is formed by end-arteries, hypotension results in ischemia and infarction in the tissues supplied by the most distal branches of these arteries. These border zones are defined by the junctions of the ACA-MCA and MCA-PCA and ACA-PCA. The classic presentation is proximal arm/leg weakness with preservation of distal strength—the "man in a barrel."
Conditions Mimicking Stroke
The diagnosis of stroke and consideration of antithrombotic or thrombolytic therapy also requires the exclusion of conditions that may mimic stroke but represent an entirely different pathophysiology.
Seizure. While seizure activity may present as a stroke, far more common are post-ictal neurologic deficits ("Todd’s paralysis"). These may manifest as any focal neurologic deficit, including weakness, sensory loss, or impaired language function. Such a deficit may last hours to days after a seizure. History of antecedent seizure is an obvious clue to the diagnosis; however, seizure may be clinically undetected or the history may be incomplete. Limb shaking does not always exclude TIA, as twitching movements have been observed in the setting of severe carotid stenosis.44
Migraine. The aura preceding a migraine headache may include focal neurological deficits such as weakness, numbness, or aphasia. Such deficits may also occur without headache, "acephalgic migraine," and these patients have a low risk of subsequent vascular event, lower than those with TIAs.45 Distinguishing migaine aura from stroke may have important implications in the acute setting if thrombolysis is a therapeutic consideration. MRI with diffusion-weighted imaging may help distinguish migraine from cerebral infarction.46
Toxic-metabolic disorders such as hypoglycemia, hyponatremia, or toxic exposures may cause focal neurologic deficits. Such insults may also "unmask" an old clinically silent stroke. Great care is required to ensure that underlying acute cerebral infarction is not also present.
Stroke—Etiology
Once the localization of a stroke is established, the evaluation should focus on an understanding of stroke pathophysiology. The 2 primary mechanisms of stroke are embolism and thrombosis. Emboli are blood clots that may originate in the heart or, more rarely, in the aortic arch. The most well-elucidated source of cardiac embolization is AF. AF is a common arrhythmia, found in 1% of people older than age 60 and more than 5% of those older than age 70.47 When associated with rheumatic heart disease and mitral stenosis, AF carries a 7-fold increased risk of stroke. The stroke risk associated with AF, though less than in rheumatic disease, is also high—5 times that of controls without AF. Overall, 20-25% of ischemic strokes are due to cardiogenic emboli.48 Almost one-half of these occur in patients with AF.49 Patients with AF who have had a previous stroke have an even higher risk of recurrence (at least 12% per year).50 Management of anticoagulation in patients with atrial fibrillation at risk for stroke will be discussed in Part II of this series.
Patients with congestive heart failure and reduced cardiac ejection fraction (EF) are at increased risk of stroke. This risk increases by approximately 18% for every 5% decrease in EF. Data from heart failure treatment studies such as Survival and Ventricular Enlargement (SAVE) suggest that warfarin may reduce stroke and mortality in patients with reduced EF, but definitive answers await specific clinical trials.51
Paradoxical emboli may originate in the venous circulation and pass into the brain via intracardiac shunts. Most shunts are congenital conditions that may only become manifest later in life because of physiologic challenges. Patent foramen ovale (PFO) is a common condition that has an increased incidence among patients with stroke. PFO may be found in as many as 40% of patients with stroke compared to 10% of the general population.52 Transesophageal echocardiography (TEE) is a powerful modality for making this diagnosis. The challenge is to prove that PFO is the etiology of stroke, rather than an incidental finding. Factors such as the size of the PFO more than 3 mm and the presence of shunting on venous injection of agitated saline increase this likelihood. Paradoxical embolism through a PFO is well documented: microbubbles have been detected in the cerebral circulation by transcranial doppler, and thrombi have actually been seen traversing a PFO.53 Arguing in favor of a possible pathogenic role, PFO is most common among patients with no identifiable cause of stroke (cryptogenic stroke). It has been detected in as many as 54% of these patients compared with only 19% of patients with an alternative explanation for their infarct.54 Patients with cryptogenic stroke and PFO also seem to have higher recurrence rates than those without PFO.55 PFO may be associated with ostium secundum atrial septal defect, which is also a stroke risk.56 Other cardiac structural abnormalities, such as mitral valve prolapse, may contribute to stroke, but a causal relationship has never been established.56
Complicated atherosclerotic plaques of the aortic arch, located proximal to the ostium of the left subclavian artery, may represent a risk for embolism to the brain. Aortic arch atherosclerosis may be implicated as a diagnosis of exclusion among ischemic stroke patients who have no significant carotid stenosis, no pattern of lacunar infarction, and no identifiable cardiac source of emboli.57 Atherosclerotic plaques, particularly those with ulceration, are common in patients older than age 60 and with vascular risk factors. Among patients with brain infarcts, ulcerated plaques were found in 21% of those between 60 to 69 years, 31% of those between 70 to 79, and 36% of those older than 80 years.58 Ulcerated plaques may promote the formation of mural thrombi, which may be exuberant, loosely adherent and mobile, and promote the formation of emboli distally to the brain or peripheral arteries. Cholesterol emboli to the brain, retina, kidney, and lower limbs may be generated from a plaque not yet covered by mural thrombus. These crystals may pass into distal arteriolar vessels and, thus, only cause very small cortical or subcortical infarctions; they are frequently asymptomatic. Cholesterol embolization may result in diffuse encephalopathy rather than focal neurological signs. Peripheral organ involvement may be helpful in the diagnosis of cholesterol emboli, especially renal infarcts, pancreatitis, intestinal infarcts, and purple (or blue) toes.59
Stenosis of the internal carotid artery (ICA) is one of the most commonly recognized sites of atherosclerosis-causing brain infarction. Ultrasound can easily localize these lesions and help identify candidates for surgical treatment. Carotid stenosis more than 70% is associated with the highest stroke risk. Part II of this series further reviews the risks and benefits of treatment of carotid stenosis.
Interestingly, patients with carotid stenosis may have other stroke mechanisms. Prospective registries, such as the Stroke Data Bank, have shown that significant high-grade stenosis of the ICA is the cause of an ipsilateral infarct of the brain in fewer than 15% of cases. In more than 20% of cases, the cause is cardioembolic, and in 25% of cases, an arteriosclerotic disease causes a lacunar stroke. Therefore, in up to 40% of cases, the exact cause of brain infarction is unknown, and by default, diagnoses such as moderate ICA stenosis (< 70%) or minor cardiac abnormalities such as PFO are accepted.60
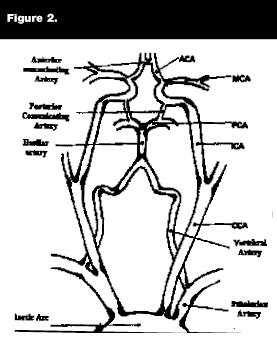
Thrombotic strokes occur as a result of in-situ clot formation, usually superimposed on atherosclerotic disease. Atherothrombotic infarcts are associated with plaques of the carotid arteries, the vertebrobasilar arteries, or the middle cerebral artery. Common areas for thrombotic occlusion are cerebral vessel branch points, as thrombosis is usually the result of clot formation that develops in the area of an ulcerated atherosclerotic plaque that occurs in an area of turbulent blood flow (see Figure 2). Patients at particularly high risk for intracranial thrombosis are patients with diabetes or of African-American descent.
Stroke in the Young
Young patients without typical risk factors for stroke or otherwise healthy older patients may still be at risk for stroke. Approximately 3-4% of all strokes occur in patients between the ages of 15 and 45 years.61 Evaluation for disease states, such as hypercoagulability, genetic syndromes, or systemic diseases posing a stroke risk, is necessary (see Table 2).
| Table 2. Stroke in the Young |
| Hypercoagulable States |
| • Protein C and S deficiency |
| • Anti-thrombin III deficiency |
| • Factor V Leiden |
| • Prothrombin gene mutation G20210A |
| • Antiphospholipid/anticardiolipin antibodies/lupus anticoagulant |
| • Sickle cell anemia |
| • Homocystinuria |
| • Pregnancy/oral contraceptive use |
| • Malignancy related hypercoagulability |
| • Polycythemia vera |
| • Thrombotic thrombocytopenic purpura |
| Vasculopathies |
| • Cervical arterial dissection |
| • Fibromuscular dysplasia |
| • Moya-moya syndrome |
| • Polyarteritis nodosa/primary CNS vasculitis |
| • Takayasu’s aortitis |
| • Vasospasm-spontaneous or related to cocaine/ amphetamines |
| • Complicated migraine |
| Cardiac Abnormalities |
| • Patent foramen ovale (PFO) |
| • Atrial septal aneurysm (ASA) |
| • Atrial myxoma |
| • Cyanotic congenital heart disease |
| Genetic Syndromes |
| • MELAS syndrome (Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) |
| • CADASIL (Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) |
| • Fabrys Disease |
| • Hereditary Hemorrhagic Telangietasias |
 |
Atherosclerosis, including large and small artery disease, is rarely the cause of stroke among patients younger than age 50. The most frequently associated factors were hypertension and hyperlipidemia, particularly hypertriglyceridemia. Any patient with familial hyperlipidemia is at increased risk for stroke, as well as for other vascular diseases such as MI.62
Carotid or vertebral artery dissection may occur spontaneously or in the setting of trauma. A dissection is produced by the subintimal penetration of blood in a cervicocephalic vessel with subsequent longitudinal extension of intramural hematoma; it is usually associated with intimal tears. Worldwide, it is probably the most common nonatherosclerotic arteriopathy. It most frequently involves the extracranial carotid, whereas intracranial vessels and both extra- and intracranial vertebral arteries are less often involved.63 The most frequent cause of neck vessel dissections is trauma, which may be mild.64 Rarely, nontraumatic movement, such as turning or flexing the head sharply, may result in dissection. Connective tissue disease type IV (Ehlers Danlos) predisposes to dissection.65 Patients with fibromuscular dysplasia are at risk for dissection, particularly young women.
The best means to diagnose arterial dissection remains conventional arteriography. Duplex ultrasound may be useful when the dissection involves the common carotid or the extracranial internal carotid artery. Magnetic resonance angiography (MRA) will probably become the gold standard, particularly with T1 axial fat saturation imaging, which allows demonstration of blood within the arterial wall (seen as a bright extraluminal crescent).66,67
In addition to PFO, other cardiac sources for stroke in young patients should be considered. Infectious, platelet-fibrin vegetations in the setting of endocarditis (SBE) may embolize. In patients with possible SBE, presenting with systemic signs or low-grade fever, anticoagulation should be withheld until this diagnosis can be ruled out. Intracerebral hemorrhage is a devastating complication aassociated with occult intracerebral mycotic aneurysms as a result of SBE.
Cardiac myxoma is a rare cause of stroke. The tumor is usually benign and originates in the left atrium in 75% of cases. It may present with constitutional symptoms, cardiac outflow obstruction, or be entirely asymptomatic. The embolic material consists primarily of myxomatous tissue, but adherent thrombotic material may also embolize. Multiple ischemic strokes, cerebral aneurysms resulting from tumor necrosis, and, rarely, intracranial hemorrage may result. Diagnosis is made by TEE; therapy is surgical.68
Stroke is a catastrophic complication of sickle cell anemia that affects 6-17% of children and young adults. Two major syndromes are observed: one results from occlusion of major cerebral vessels and primarily affects children from 2 to 15 years of age (mean, 6-7 years).69,70 The other results from intracerebral or subarachnoid hemorrhage and affects older children and adults. The risk of stroke appears to be increased in patients with HbF levels of less than 8% and in patients with siblings who have suffered a stroke. Risk of stroke related to sickle cell anemia may be assessed using transcranial doppler ultrasound.71
Antiphospholipid antibodies (APLA) are a group of antibodies directed against cell surface phospholipids. The frequency of APLA depends heavily on the population being studied. APLAs are detected in 10-26% of patients with first stroke and 40-50% of patients with systemic lupus erythematosus. APLAs are often related to a number of concomitant risk factors for thrombosis such as pregnancy, hypertension, hyperlipidemia, and diabetes. APLAs are associated with other neurological disease such as cerebral or spinal arterial or venous infarction, chorea, hemidystonia, seizures, migraine, Guillain-Barré syndrome, transient global amnesia, motor neuron disease, myasthenia gravis, and behavioral abnormalities such as affective disorder or dementia.72 Patients with APLAs and cerebral thrombosis face a recurrence risk up to 20% per year for TIA or stroke and 56% per year for noncerebral thrombosis.73
Sneddon Syndrome is a rare pathology, more frequent in women, characterized by stroke, livedo reticularis, and antiphospholipid antibodies.74 Hypertension may be present. Livedo reticularis may be present several years before the onset of neurologic symptoms. Cigarette smoking and the use of oral contraceptives are frequently found in conjunction with Sneddon syndrome, but the relationship between these risk factors and the disease is not clearly established.75
Multiple other factors may contribute to a hypercoagulable state (see Table 2). Several studies suggest that protein C or S deficiency may be a cofactor in the etiology of stroke.76 Pregnancy and the use of oral contraceptives are significant risk factors for hypercoagulability and the development of stroke.77
Homocystinuria is characterized by the increase of homocysteine and methionine in blood and urine. The inherited form corresponds to a decrease in cystathione-b-synthase activity, which is an enzyme crucial for the transfer of the sulfation of cysteine from methionine.78 This results in an increase in both blood and urine levels of methionine and homocysteine. Both homozygotes and heterozygotes have an increased risk for stroke resulting from early atherosclerosis. Patients with non-genetic increases in homocysteine levels are also at risk for atherosclerosis.79
Vasculitis may cause stroke in the setting of primary CNS disease or as part of a systemic syndrome such as systemic lupus erythematosis. Vasculitis causes focal or multifocal cerebral ischemia through inflammation and necrosis of extracranial and/or intracranial blood vessels and through bland vasculopathic changes. The diagnosis of CNS vasculitis is aided by the presence or absence of peripheral nervous system or systemic organ involvement and by identifying the underlying cause of the inflammation. Primary CNS vasculitis, Behcet’s disease, Takayasu’s arteritis, and temporal arteritis are notable for their infrequent involvement or complete lack of involvement of the peripheral nervous system. By contrast, the hypersensitivity and systemic necrotizing vasculitides frequently produce polyneuropathy.
Focal cerebral vasoconstriction may be the consequence of a reversible segmental narrowing due to sympathomimetic drugs such as ergot derivatives, crack cocaine, methylamphetamine, and phenylpropanolamine. The latter has recently been removed from over-the-counter cold preparations. In the idiopathic form, spontaneous vasoconstriction may occur in adults without risk factors for stroke. The spontaneous form is more common in women and among those with a history of migraine.80,81
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) is characterized by recurrent subcortical ischemic stroke and dementia. Moya-moya disease is a clinical entity characterized by bilateral cerebral infarcts due to bilateral hypoplasia of the internal carotid arteries. The term derives from the Japanese word for "puff of smoke" and was proposed because of the unusual feathery appearance of the vascular network found in these patients as seen on angiography. It affects children most frequently, with a maximal peak in frequency older than the age of 6 years. Clinical presentation is varied, and repeated TIAs can be observed, mainly at the onset of the disease.82
References
1. American Heart Association: Heart and Stroke Facts Statistics: 1997 Statistical Supplement. American Heart Association. Dallas, Tex; 1997.
2. Matchar DB, Duncan PW. Cost of stroke. Stroke Clinical Updates. 1994;5:9-12.
3. Wentworth DA, Atkinson RP. Implementation of an acute stroke program decreases hospitalization costs and length of stay. Stroke. 1996;27:1040-1043.
4. Taylor TN, et al. Lifetime cost of stroke in the United States. Stroke. 1996;27:1459-1466.
5. Sacco RL, et al. Risk factors panel: American Heart Association Prevention Conference IV: Prevention and rehabilitation of stroke: Risk factors. Stroke. 1997;28:1507-1517.
6. Albers GW, et al. Supplement to the guidelines for the management of transient ischemic attacks. Stroke. 1999; 30:2502-2511.
7. The sixth report of the joint National Committee on Prevention, Detection, Evaluation and Treatment of high blood pressure. Arch Intern Med. 1997;157:2413-2446.
8. Brown RD, et al. Stroke incidence, prevalence and survival: Secular trends in Rochester, Minnesota, through 1989. Stroke. 1996; 27:373-380.
9. Burt VL, et al. Prevalence of hypertension in the US adult population: results from the third National Health and Nutrition Examination Survey, 1988-1991. Hypertension. 1995;25:305-313.
10. Collins R, et al. Blood pressure, stroke, and coronary heart disease, part 2: Short term reductions in blood pressure: Overview of randomised drug trials in their epidemiological context. Lancet. 1990;335:827-838.
11. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991;265:3255-3264.
12. Wolf PA, et al. Probability of stroke: A risk profile from the Framingham Study. Stroke. 1991;22:312-318.
13. Kuller LH, Dorman JS, Wolf PA. Cerebrovascular diseases and diabetes. In: National Diabetes Data Group Department of Health and Human Services, National Institutes of Health, ed. Diabetes in America: Diabetes Data Compiled for 1984. Bethesda, Md: National Institutes of Health; 1985:1-18.
14. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977-986.
15. Demchuk AM, et al. Serum glucose level and diabetes predict tissue plasminogen activator-related intracerebral hemorrhage in acute ischemic stroke. Stroke. 1999;30:34-39.
16. Abbott RD, et al. Risk of stroke in male cigarette smokers. N Engl J Med. 1986;315:717-720.
17. Bonita R, et al. Cigarette smoking and risk of premature stroke in men and women. BMJ. 1986;293:6-8.
18. Howard G, et al, for the The Atherosclerosis Risk in Communities (ARIC) Study. Cigarette smoking and progression of athero-sclerosis. JAMA. 1998;279:119-124.
19. Sacks FM, et al. The effect of pravastatin on coronary events. A comparison of two doses of aspirin (30 mg vs 283 mg a day) in patients after a transient ischemic attack or minor after myocardial infarction in patients with average cholesterol levels: Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001-1009.
20. Hebert PR, et al. Cholesterol lowering with statin drugs, risk of stroke, and total mortality: An overview of randomized trials. JAMA. 1997;278:313-321.
21. Donahue RP, et al. Alcohol and hemorrhagic stroke: The Honolulu Heart Program. JAMA. 1986;255:2311-2314.
22. Gill JS, et al. Stroke and alcohol consumption. N Engl J Med. 1986;315:1041-1046.
23. Pellegrini N, et al. Effects of moderate consumption of red wine on platelet aggregation and haemostatic variables in healthy volunteers. Eur J Clin Nutr. 1996;50:209-213.
24. Selhub J, et al. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med. 1995;332:286-291.
25. Giles WH, et al. Total homocyst(e)ine concentration and the likelihood of nonfatal stroke: results from the Third National Health and Nutrition Examination Survey, 1988-1994. Stroke. 1998;29:2473-2477.
26. Boushey CJ, et al. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. JAMA. 1995;274:1049-1057.
27. Fletcher GF. Exercise in the prevention of stroke. Health Rep. 1994;6:106-110.
28. Viscoli CM, et al. Estrogen after ischemic stroke: Effect of estrogen replacement on risk of recurrent stroke and death in the Women’s Estrogen for Stroke Trial. Stroke. 2001;32:329.
29. Preventive Services Task Force. Guide to Clinical Preventive Services: Report of the U.S. Preventive Services Task Force. Baltimore, Md: Williams & Wilkins; 1996: xcii, 953.
30. Broderick J, et al. The greater Cincinnati/Northern Kentucky stroke study. Stroke. 1998;29:415-421.
31. Williams GR, et al. Incidence and occurrence of total (first-ever and recurrent) stroke. Stroke. 1999;30: 2523-2528.
32. Brainin M, et al. Silent brain infarcts and transient ischemic attacks. Stroke. 1995;26:1348-1352.
33. Wolf PA, et al. Preventing ischemic stroke in patients with prior stroke and transient ischemic attack. Stroke. 1994; 25:1901-1914.
34. Levy DE. How transient are transient ischemic attacks? Neurology. 1988;38:674-677.
35. Whisnant JP, et al. Transient cerebral ischemic attacks in a community: Rochester, Minnesota, 1955 through 1969. Mayo Clin Proc. 1973;48:194-198.
36. Dennis M, et al. Prognosis of transient ischemic attacks in the Oxfordshire Community Stroke Project. Stroke. 1990;21:848-853.
37. Johnston SC, et al. Short-term prognosis after emergency department diagnosis of TIA. JAMA. 2000;284:2901-2906.
38. Kidwell CS, et al. Diffusion MRI in patients with transient ischemic attacks. Stroke. 1999;30:1174-1180.
39. Fisher CM. Lacunar strokes and infarcts: A review. Neurology. 1982;32:871.
40. Sacco SE, et al. Epidemiological characteristics of lacunar infarcts in a population. Stroke. 1991;22:1236-1241.
41. Nadeau SE, et al. Stroke rates in patients with lacunar and large-vessel celebral infarctions. J Neurol Sci. 1993; 114:128-137.
42. Millikan C, Futrell N. The fallacy of the lacune hypothesis. Stroke. 1990;21:1251-1257.
43. Segal AZ, Rordorf G. Stroke as a complication of cardiac catheterization. Neurology. 2001; In press.
44. Baquis GD, Pessin MS, Scott RM. Limb shaking—a carotid TIA. Stroke. 1985;16:444-448.
45. Dennis M, Warlor C. Migraine aura without headache: Transient ischaemic attack or not? J Neurol Neurosurg Psychiatry. 1992;55:437-440.
46. Cutrer FM, et al. Perfusion-weighted imaging defects during spontaneous migrainous aura. Ann Neurol 1998;43(1):25-31.
47. The National Heart Lung and Blood Institute Working Group on Atrial Fibrillation: Atrial fibrillation: Current understandings and research imperatives. J Am Coll Cardiol. 1993;22:1830.
48. The Stroke Prevention in Atrial Fibrillation Investigators: Predictors of thromboembolism in atrial fibrillation: I. Clinical features of patients at risk. Ann Intern Med. 1992;116:1.
49. EAFT (European Atrial Fibrillation Trial) Study Group: Secondary prevention in non-rheumatic atrial fibrillation after transient ischemic attack or minor stroke. Lancet. 1993;342:1255.
50. Wolf PA, et al. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke. 1991;22:983-988.
51. Pullicino PM, et al. Stroke in patients with heart failure and reduced left ventricular ejection fraction. Neurology. 2000;54: 288-294.
52. Hausmann D, et al. Diagnosis of patent foramen ovale by transesophageal echocardiography and association with cerebral and peripheral embolic events. Am J Cardiol. 1992;70:668-672.
53. Nelson CW, et al. Impending paradoxical embolism: Echocardiographic diagnosis of an intracardiac thrombus crossing a patent foramen ovale. Am Heart J. 1991;122:859-862.
54. Nelson CW, et al. Impending paradoxical embolism: Echocardiographic diagnosis of an intracardiac thrombus crossing a patent foramen ovale. Am Heart J. 1991;122:859-862.
55. Lechat P, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148-1152.
56. Kichura GM, Castello R. Abnormalities of the interatrial septum as a potential cardiac source of embolism: Patent foramen ovale and atrial septal aneurysm. Echocardiography. 1993;10:441.
57. Barnett JHM, et al. Further evidence relating mitral valve prolapse to cerebral ischemic events. N Engl J Med. 1980;302:139-144.
58. Amarenco P, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331:1474-1479.
59. Amarenco P, et al. The prevalence of ulcerated plaques in the aortic arch in patients with stroke. N Engl J Med. 1992;326:221-224.
60. Ezzeddine MA, et al. Clinical characteristics of pathologically proved cholesterol emboli to the brain. Neurology. 2000;54(8):1681-1683.
61. Sacco RL, et al. Infarcts of undetermined cause: The NINCDS stroke data bank. Ann Neurol. 1989;25:382-390.
62. Hachinski VE, Norris JW. The young stroke. In: Davis FA, ed. The Acute Stroke. Philadelphia, Pa; 1985.
63. Bansal BC, et al. Familial hyperlipidemia in stroke in the young. Stroke. 1986;17:1142-1145.
64. Biller J, et al. Cervicocephalic arterial dissection: A ten-year experience. Arch Neurol. 1986;43:1234-1238.
65. Tramo MJ, et al. Vertebral injury and cerebellar stroke while swimming: Case report. Stroke. 1985;16:1039-1042.
66. Schievink WI, et al. Cerebrovascular disease in Ehlers-Danlos syndrome type IV. Stroke. 1990;21:626-632.
67. Hiserman JE, et al. MR angiography of cervical fibromuscular dysplasia. AJNR Am J Neuroradiol. 1992;13:1454-1457.
68. Knepper LE, et al. Neurologic manifestations of atrial myxoma. Stroke. 1988;19:1435-1440.
69. Sarnaik SA, Lusher JM. Neurological complications of sickle cell anemia. Am J Pediatr Hematol Oncol. 1982;4:386-394.
70. Van Hoff J, et al. Intracranial hemorrhage in children with sickle cell disease. Am J Dis Child. 1985;139:1120-1123.
71. Adams RJ, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11.
72. Gorman DG, Cummings JL. Neurobehavioral presentations of the antiphospholipid antibody syndrome. J Neuropsychiatry Clin Neurosci. 1993;5:37-42.
73. Feldmann E, Levine SR. Cerebrovascular disease with antiphospholipid antibodies: immune mechanisms, significance, and therapeutic options. Ann Neurol. 1995;37(Suppl 1):S114-130.
74. Montalban J, et al. Sneddon’s syndrome and anticardiolipin antibodies. Stroke. 1988;19:785-786.
75. Kalashnikova LA, et al. Sneddon’s syndrome and the primary antiphospholipid syndrome. Cerebrovasc Dis. 1994;4:76.
76. Allaart CF, et al. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet. 1993;341:134-138.
77. Wiebors . Ischaemia cerebrovascular complications of pregnancy. Arch Neurol. 1985;42:1106-1113.
78. Mudd SH, et al. The natural history of homocystinuria due to cystationine-b-synthase deficiency. Am J Hum Genet. 1985;37:1-31.
79. Boers JH. Hyperhomocystinemia: Newly recognized risk factor for vascular disease. Neth J Med. 1994;45:34-41.
80. Bogousslavsky J, et al. Migraine stroke. Neurology. 1988;38:804-806.
81. Call Gk, et al. Revesible cerebral segmental vasoconstriction. Stroke. 1988;19:1159-1170.
82. Bruno A, et al. Celebral infarction due to moyamoya disease in young adults. Stroke. 1988;19:826-833.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.