Antibiotic Resistance and the Problem of Antibiotic Overuse
Antibiotic Resistance and the Problem of Antibiotic Overuse
Author: James A. Wilde, MD, FAAP, Assistant Professor of Emergency Medicine, Section of Pediatric Emergency Medicine, Medical College of Georgia, Augusta.
Peer Reviewer: David Karras, MD, FACEP, FAAEM, Associate Professor of Medicine, Director of Emergency Medicine Research, Temple University School of Medicine, Philadelphia, PA.
Antibiotics were introduced more than 60 years ago and have been a major boon to health care. At the beginning of this century, tuberculosis (TB) and pneumonia were the leading causes of death in the United States. Together with diarrheal disease, they accounted for 30% of all deaths. Between 1938 and 1952, the early years of the antibiotic era, deaths from infectious disease in the United States declined at an annual rate of 8.2%. By 1997, pneumonia/influenza was the lone infectious disease among the top 10 causes of death in the United States, ranking only sixth.1 There are now more than 100 antibiotics available for medical use, and no Western-trained physician would consider trying to practice medicine without these powerful therapeutic allies. Despite their unquestioned value in treating patients with bacterial infections, in recent years there have been increasing warnings about their use.2-6 These warnings focus on the potential for overuse, which may cause bacterial resistance to the killing effect of the antibiotic. Consequences may include a greater likelihood for hospitalization, longer hospital stays, and increased mortality.7 The problem is global in its scope, prompting a major international meeting in Geneva, Switzerland, in 1995. The findings from this meeting were published in extensive detail in a supplement to Clinical Infectious Diseases in 1997.8 On the national level, extensive effort is expended through the National Antimicrobial Resistance Monitoring System and the National Nosocomial Infections Surveillance System to study resistance in the United States and to recommend means to slow its inexorable spread.
In this article, the author examines antibiotic resistance, including a brief review of the mechanism of action of certain key antibiotics, mechanisms of resistance, resistance patterns, strategies for reducing resistance, and recommendations for reducing antibiotic use in treating illnesses for which they are not indicated.
— The Editor
Historical Perspective
Studies of bacteria isolated from closed systems from the pre-antibiotic era have shown that prior to the introduction of antibiotics, there were few genes coding for antibiotic resistance.9 Staphylococcus aureus uniformly was sensitive to penicillin at the time of its release in 1941. Tables 1 and 2, which compare beta-lactam antibiotic development and the emergence of resistance, reveal the problem that emergency medicine physicians face: As fast as new antibiotics are developed, the microbes find a way to thwart them. Penicillin resistant S. aureus was recognized within one year of the introduction of penicillin.10 Rapid expansion of penicillin resistance among staphylococcus isolates around the world led to a crisis in the 1950s, a crisis that appeared to be solved with the introduction of methicillin, a penicillinase-resistant penicillin, in 1961. However, just one year later, methicillin resistant isolates were identified.11 Similarly, the 1963 development of ampicillin, an extended spectrum penicillin, was considered to be a major step forward in the treatment of gram-negative bacterial infection; however, later that same year Escherichia coli isolates in Greece were shown to produce plasmid-mediated TEM-1 beta-lactamase.12 The introduction in 1964 of cephalosporins, an entirely new class of antibiotics, gave the medical world a powerful new ally, one that was much less susceptible to beta-lactamases. Soon after, resistance to cephalosporins became widespread, and by the 1970s gram-negative bacilli became the predominant nosocomial pathogens.13 Strains of Klebsiella oxytoca that were resistant to cefuroxime appeared within a year of its introduction in 1978.14 In 1983, extended spectrum beta-lactamases were first reported, only two years after the release of cefotaxime.15 Resistance to imipenem, from the new class of carbapenims, was discovered in 1982, three years before its release in the United States.16 To date, more than 100 resistance genes have been identified among gram-positive and gram-negative bacteria, and that number is increasing rapidly.17
| Table 1. Antibiotic Development |
| Beta-Lactams |
| • Penicillin |
| • Methicillin |
| • Ampicillin |
| • Cephalothin |
| • Cefoxitin |
| • Cefotaxime |
| • Amoxicillin/Clavulanate |
| • Imipenem |
| • Aztreonam |
| Table 2. The "Bugs" Strike Back |
| 1942: Penicillin-resistant S. aureus reported. |
| 1961: Methicillin-resistant S. aureus reported. |
| 1963: Plasmid mediated TEM-1 beta-lactamase resistance described. |
| 1978: Cefuroxime-resistant Klebsiella oxytoca reported. |
| 1982: Carbapenemase isolated from Serratia. |
| 1983: Extended-spectrum beta-lactamases reported. |
One of the lessons of the past 60 years is that as fast as we develop antibiotics, the bacteria develop resistance. Part of the explanation for this is that the microbes have us vastly outnumbered. We may kill billions of bacteria during a single course of antibiotics, but if even one mutant organism exists in that patient, the possibility exists for the rapid establishment of a resistant population. Some bacteria can replicate themselves as rapidly as every 20 minutes. In just eight hours, a bacterium with this doubling time can number well into the millions. In one recent study, volunteers were given orally administered teicoplanin for 21 days. While no glycopeptide resistance was noted at baseline, 81% of the volunteers developed vancomycin-resistant enterococcus (VRE) during the study, with colony counts as high as 108 colony forming units per gram of stool.18 With the notable exception of group A streptococcus, which is still uniformly sensitive to penicillin despite 60 years of widespread use,19 it appears that most bacteria will eventually develop resistance to a specific antibiotic if given enough exposure.
The struggle between man and microbe can be compared to a war of attrition, but in this war "technology is losing the arms race with evolution."4 We can postpone our day of defeat by using our resources wisely. Unfortunately, the rate of attrition has been increased by some physicians who are indiscriminate in their use of antibiotics, and by liberal policies in some countries that allow the public to buy antibiotics without a prescription or a doctor’s guidance. Every time we use an antibiotic inappropriately, such as during an obvious viral infection, all of the billions of bacteria in the nasopharynx and gastrointestinal tract are exposed to the antibiotic simultaneously, and sensitive strains will be killed. Even the excretion of antibiotics in sweat can affect the antibiotic susceptibility of bacteria on skin.20 The obvious result is that resistant bacteria can then expand their numbers.
Inappropriate Antibiotics
Inappropriate antibiotic use among licensed physicians in the United States and elsewhere is well documented.21-24 Data derived from the National Ambulatory Medical Care Survey, a survey of office-based physicians, show that antibiotics are routinely prescribed for colds (51%), upper respiratory tract infections (52%), and bronchitis (66%) for patients older than 18 years.22 In an accompanying editorial, it is further noted that more than three-fourths of the antimicrobial drug prescriptions written annually in physicians’ offices are given to people with respiratory infections.3 While some of these antibiotics may be warranted, particularly for bacterial superinfection, most are not. Another study in the pediatric literature showed similar overuse.23 Several case management vignettes were presented to pediatricians and family practitioners in Northern Virginia. Seventy-one percent of family practitioners and 53% of pediatricians prescribed antibiotics in infants with new onset upper respiratory infection and green nasal secretions. Further, only 15% of family practitioners and 23% of pediatricians waited for 7-10 days of persistent purulent nasal discharge before prescribing antibiotics, despite published guidelines recommending antibiotics only for purulent nasal discharge of longer than 10-14 days.25 It has been estimated that the rate of sinusitis diagnosis in the United States exceeds the actual predicted incidence by at least ten-fold.26
Physicians often prescribe antimicrobials early in the course of a cold in an attempt to prevent bacterial complications such as pneumonia. This strategy has been found to be of no value in otherwise healthy children.27 Other studies examining the role of episodic antibiotics in preventing otitis media during colds have shown mixed results, and this strategy is, at best, less effective than standard continuous prophylaxis.25,28 Furthermore, even in patients at high risk for recurrent otitis media, continuous antibiotic prophylaxis has limited efficacy, and some experts have begun to suggest that its use should be severely curtailed in favor of other options such as elimination of smoking in the home, tympanostomy tube placement, influenza vaccination, or treatment only for episodes of recurrent infection.28,29
Some physicians argue that antibiotics can’t do any harm, and may be helpful, so why not use them? There are ever-increasing reasons to refute this argument. First, antibiotics are expensive, particularly the cephalosporins and newer macrolides. Millions of unnecessary antibiotic prescriptions can be equated with many more millions of wasted health care dollars. Second, there are potential side effects from the administration of any antibiotic, such as allergic reactions, diarrhea, abdominal discomfort, vomiting, and fungal overgrowth leading to oral thrush or candidal diaper rash.30 Third, the administration of antibiotics is unlikely to alter the course of a viral illness, but parental expectations after their still-febrile child has been on an antibiotic for two or three days may lead to further unnecessary health care visits and additional prescriptions for more antibiotics. Fourth, patients taking even short courses of antibiotics are at significant risk for the development of resistant organisms.31 In addition, antibiotics affect not only the patient taking them, but also those with whom they come into contact. Children in schools and day care centers who have taken no antibiotics may still acquire resistant organisms from their classmates through horizontal transfer.32 Finally, there are new reports of potentially serious consequences of antibiotic use. Antibiotics taken during a bout of diarrhea may increase the risk of hemolytic-uremic syndrome, specifically when trimethoprim-sulfamethoxazole is used in a patient with Escherichia coli 0157:H7 enteritis.33 Other studies have shown a correlation between the administration of antibiotics and the subsequent development of asthma.34-35
Another explanation given by some physicians for liberal antibiotic prescribing practices is parental pressure and concern over patient satisfaction.36 They reason that if antibiotic-seeking patients comes to their offices, they will lose those patients as customers if this agenda is not met. While the literature on this topic is divided, several studies in ambulatory care settings have shown that receipt of an antibiotic is not as important to these patients as a thorough explanation from the physician about the illness.37,38
Key Concepts
General Mechanisms of Resistance. A number of specific mechanisms to resist the action of antibiotics have evolved within the microbial world. It is helpful to consider three different, broad strategies that have evolved before examining the specific mechanisms.
First, the antibiotic can be altered in some way. A common theme in biology is the interaction between an effector molecule, such as a hormone or an antibody, and its target binding site, which is usually a complex molecule as well. This involves complex interactions at the molecular level, which in turn rely on proper orientation of the molecules. If one of the molecules is altered in such a way that the tertiary structure also is changed, binding may be prevented, and the biologic effect is prevented. The same principle can be applied to antibiotics, which usually have a specific binding site. If the bacterium can produce an enzyme that alters the antibiotic, it also may alter the tertiary structure of the antibiotic enough to prevent it from attaching to its binding site. A well-recognized example of this strategy is the development of beta-lactamases, which alter beta-lactam antibiotics and render them unable to exert their antibacterial effect.39
Second, the binding site of the antibiotic can be altered. Most antibiotics must bind to a specific site in or on the bacterium to exert their effect. These binding sites are antibiotic specific, so different binding sites are used for penicillins, aminoglycosides, glycopeptides, sulfonamides, and quinolones. If the binding site is altered in some way to make it unrecognizable to the antibiotic, binding does not occur and the antibiotic loses its effect. This is the primary mechanism used by Streptococcus pneumoniae in eluding the effect of penicillin.40
Third, bacteria may develop ways to deny the antibiotic access to the binding site. Many antibiotics (i.e., aminoglycosides and quinolones) must gain access to the inside of the bacterial cell to attach to their binding site. Access is accomplished through the use of porins, an opening in the cell membrane that allows certain materials to pass from the extracellular environment to the inside of the cell. If these porins are altered in such a way that the antibiotic cannot get inside the cell, there is no way for the antibiotic to reach its binding site and the antibiotic effect cannot be achieved. Permeability mutants are well described among gram-negative bacteria resistant to quinolones or to aminoglycosides.41 In addition, many gram-negative organisms have an intrinsic resistance to many antibiotics due to the presence of efflux pumps that actively remove antimicrobial agents from the intracellular environment.42
Spread of Resistance. The development of resistance in individual populations of bacteria within a patient is only the beginning of the problem. After the development of resistant strains of bacteria, the problem is further compounded by the spread of resistance to other patients or to other populations of bacteria. It is this spread that magnifies antibiotic resistance and makes it a community-wide problem. Again, the microbes have developed an impressive array of mechanisms by which they can spread.
The simplest mechanism for the spread of resistance is the selection of naturally resistant mutants. In some bacterial populations, there exists a background rate of natural resistance to a specific antibiotic, conferred solely by chance. Usually these background rates are quite low, perhaps no more common than one in 106 or 108 organisms. If the bacterial population is exposed to that antibiotic, most of the bacteria will be susceptible and will be killed. However, small numbers of non-susceptible bacteria may remain and multiply to occupy the niche formerly occupied by their susceptible comrades. Broad use of this antibiotic could lead to the development of resistant populations of organisms in many different patients. In most bacterial infections, the background rate of resistance is low, the bacterial load is relatively low, and any remaining non-susceptible organisms can be eliminated by the patient’s own immune system. In these infections, development of naturally resistant mutant populations is not likely. If the bacterial load is very high, or the bacteria are able to elude the patient’s immune system, development of resistant populations becomes more likely. An example of bacteria with a background rate of natural resistance to antibiotics is Mycobacterium tuberculosis (MTB).43
Another simple method for amplification of resistance is clonal spread. Once a resistant population is established in an individual, contamination of the environment with those resistant bacteria can occur. If another person comes in contact with that contaminated environment, the resistant bacteria can spread to that unaffected person, and then multiply enough to be of clinical significance. This mechanism is probably of importance in the spread of VRE and in methicillin-resistant Staphylococcus aureus (MRSA) infections.44 This also is the mechanism for the spread of most infectious diseases in the world, such as the common cold, influenza, or varicella.
Several other mechanisms of spread utilize the sharing of nucleic acid between bacteria. The principal example of this mechanism is via plasmids, circular bands of DNA that are present in many bacteria. This nucleic acid is extra-chromosomal; that is, it is not a natural part of the genome, and exists separately from the chromosomes themselves. Plasmids can code for a number of proteins, including beta-lactamases and other antibiotic resistant gene products. Plasmids can be transferred from one bacterium to another within a species, or even from one species of bacteria to another. In this way, resistant genes may appear in a bacterial population even though that population has never encountered the antibiotic.
Another mechanism for bacteria to exchange genetic material is through transposons. Transposons are segments of DNA that can move from chromosome to plasmid within the same bacterium or from a chromosome in one bacterium to a chromosome in another. Thus resistance genes can be transferred directly from the chromosome of a resistant bacterium to the chromosome or a plasmid of another bacterium, in a manner similar to the spread of plasmids. Again, this can result in the development of resistant organisms that have never encountered the antibiotic to which they are resistant.
A final, insidious method for spread of resistance is through co-selection with other resistance genes. Many plasmids contain multiple genes coding for resistance to more than one antibiotic.45 Consider a plasmid that contains resistance genes for both sulfonamides and penicillin. If that plasmid is widely distributed in a patient who receives penicillin therapy, all the isolates without the plasmid will be killed and all the isolates with the plasmid will survive and multiply. But because the plasmid also contains the gene for sulfonamide resistance, the resulting population of bacteria also will be uniformly resistant to sulfonamides, even though no sulfonamides have been used in the patient. Some plasmids can contain multiple genes for resistance, rendering those organisms resistant to an entire array of antibiotics. Linked resistance genes on plasmids are a major reason for the common occurrence of multiply resistant gram-negative enteric bacteria.45
Means to Retard Dissemination of Resistance. There are only a limited number of options available to slow down the relentless increase of bacteria that are resistant to antibiotics. Chief among them is limiting the use of antibiotics, so that the emergence of resistance can be delayed. The more frequently a bacterial population is exposed to a specific antibiotic, the more likely it is that resistance will emerge. Conversely, with less frequent exposure of the bacteria to the antibiotic, there is less likelihood of resistance.46
Infection control measures also can be helpful for decreasing the spread of resistance. In essence, this is analogous to decreasing the spread of infectious diseases themselves. Quarantining of infected patients can decrease the spread of diseases such as TB, diphtheria, or cholera by decreasing the contamination of uninfected people. Likewise, the use of isolation rooms, gowns, gloves, and dedicated stethoscopes can decrease the spread of VRE or MRSA from an infected individual.44,47
A third method to reduce resistance, which often is ignored, is to use antimicrobials with the least selective effect on the patient’s bacterial flora. Therapy for impetigo should be directed toward group A streptococci and coagulase-positive staphylococci, the primary etiologic agents. Appropriate oral antibiotics include erythromycin, clindamycin, or cephalexin. The use of a more powerful antibiotic, such as an extended spectrum cephalosporin, gains nothing for the patient. The primary potential advantage of extended spectrum cephalosporins is that they kill a broader array of organisms, including many gram-negative organisms. They will treat impetigo, but they also will kill many other bacterial inhabitants of the nasopharynx and the gastrointestinal tract that are not targets of therapy. In general, when therapy is directed toward a known pathogen, the narrowest spectrum antibiotic that will kill the target organism is the best choice for the patient and for the community, since this results in less selective pressure on other bacteria in the environment.18
Selected, Specific Resistance Mechanisms
Beta-Lactam Antibiotics. The beta-lactam ring is a structure common to all penicillins, cephalosporins, and monobactams, and its integrity is essential for antibacterial activity. (See Figure 1.) The target of a beta-lactam antibiotic is one of a number of penicillin-binding proteins (PBP). A PBP is a cell membrane bound peptidase present in bacteria but not present in human cells. This peptidase aids in cell wall cross bridge formation, a step that is required for the structural integrity of the growing bacterial cell wall. When a beta-lactam antibiotic binds to a PBP, proper cell wall formation is prevented, leading to cell death. There can be several different PBPs in the same bacterium.
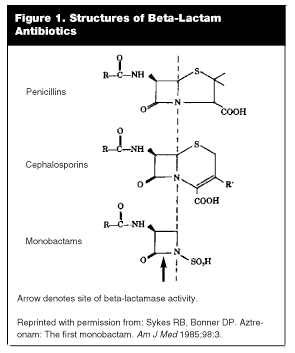 |
A beta-lactamase is an enzyme (protein) produced by the bacterium that cleaves to a specific region of the beta-lactam ring. The resulting alteration in the structure of the antibiotic molecule causes it to lose its ability to interfere with bacterial replication, rendering it useless as an antibacterial agent. More than 170 different beta-lactamases have been identified to date, a number that has doubled in the last decade.39 Beta-lactamases share several highly conserved amino acid sequences with PBPs. The beta-lactamase protein can be thought of as a decoy protein; in essence, the beta-lactam antibiotic is "tricked" into binding to the beta-lactamase molecule instead of the PBP. Once binding has occurred, the beta-lactamase hydrolyzes the beta-lactam ring, inactivating the antibiotic.
Genes present in the chromosome or on plasmids can control production of beta-lactamases. Transposons encode at least 13 plasmid determined beta-lactamases and two extended spectrum beta-lactamases.39 There frequently are multiple beta-lactamases present in a single organism. Beta-lactamases are grouped into four classes: A, B, C, and D. Some gram-positive organisms, such as S. aureus, can secrete class A beta-lactamase outside the cell, providing population resistance to other organisms in the immediate vicinity. This mechanism may explain the failure of penicillin therapy in some cases of group A streptococcus pharyngitis.48 Almost all gram-negative bacteria produce Class C beta-lactamase, which virtually always remains cell-bound. Class C beta-lactamase is produced at low, clinically insignificant levels in the absence of antibiotics. However, in the presence of beta-lactam antibiotics, the genes controlling its production can be induced, leading to hyperproduction of the Class C beta-lactamase and highly resistant organisms. Inducible production of beta-lactamase is particularly common among isolates of Enterobacter cloacae after exposure to third-generation beta-lactam antibiotics.39
In a February 1999 study of nursing home patients hospitalized for sepsis due to E. coli or Klebsiella, molecular fingerprinting showed seven strain types circulating in this population. Closer inspection revealed that 17 of 20 isolates carried the same plasmid, which coded for a relatively rare beta-lactamase. This suggests that the outbreak was caused not by clonal spread of a specific strain of bacteria, but by spread of the plasmid itself from one strain of bacteria to others. This plasmid conferred resistance to ceftazidime, gentamicin, and tobramycin.49
A second mechanism conferring resistance to beta-lactams is alteration of PBPs, as discussed above. This is the mechanism used by S. pneumoniae, which explains why use of a beta-lactamase resistant antibiotic does not overcome penicillin resistance among pneumococci. Some isolates of S. pneumoniae are intermediately resistant to penicillin, but this low-level resistance can be overcome by increasing the concentration of the antibiotic. This finding has led to recent recommendations for the treatment of otitis media with high-dose amoxicillin in those patients who have failed standard therapy.50
Finally, some resistance to beta-lactams is mediated through alterations in the permeability of the bacterial cell membrane. Porin deficient mutants of E. cloacae were first recognized in 1985. This porin deficiency conferred resistance to imipenem due to the inability of the antibiotic to penetrate to the active site in sufficient quantities.51
Vancomycin. Vancomycin and teicoplanin are related glycopeptide antibiotics. They have a unique spectrum of activity in that they are effective only against gram-positive organisms. They are used primarily as second-line therapy against penicillin- or cephalosporin-resistant organisms such as staphylococci or enterococci. Like the beta-lactam antibiotics, glycopeptides interfere with bacterial cell wall synthesis, leading to cell death, but the binding site is unique. The target for glycopeptides is the D-alanine-D-alanine terminal of peptidoglycan. When vancomycin binds, the peptidoglycan terminal is unavailable for the peptidase that catalyzes the formation of peptidoglycan cross bridges. Recall that beta-lactam antibiotics bind the peptidase itself. Thus, beta-lactams and vancomycin both block cross-bridge formation, one by binding the peptidase, the other by binding the target of the peptidase.
Resistance is mediated through Van proteins found in the cytoplasm of resistant organisms. Van proteins alter the peptidoglycan terminal from D-ala-D-ala to D-ala-D-lactate. This alteration results in the inability of vancomycin to bind, but does not alter the ability of the organism to form cross bridges.
Three different resistance phenotypes have been described: VanA, VanB, and VanC. Expression of VanA and VanB is inducible, leading to very high levels of resistance to vancomycin and teicoplanin in the case of VanA, and resistance to vancomycin alone in the case of VanB. VanA usually is carried and transferred on plasmids. VanB is transferable by conjugation.52 The resistance genes for VanC are probably chromosomal and are not inducible.53
Aminoglycosides. The target of aminoglycosides is the 30S bacterial ribosome. Ribosomes that are bound in this manner are unable to carry out the protein synthetic functions for which they are essential, leading to cell death. Mammalian ribosomes differ substantially from bacterial ribosomes, and are, therefore, unaffected by aminoglycosides.
More than 50 different aminoglycoside-modifying enzymes have been described.41 These enzymes can change the aminoglycoside molecule in a manner analogous to the changes induced in beta-lactam antibiotics by beta-lactamases. The resulting alterations can interfere with the binding of the antibiotic to its target, leading to resistance. These modifying enzymes can be mediated by either chromosomes or plasmids, thus providing ample opportunities for further spread of the resistance genes through conjugation as previously discussed. Resistance to aminoglycosides due to alterations in permeability also has been described.
Quinolones. The bacterial target for quinolones is DNA gyrase or topoisomerase, both of which are vital in cell replication. In essence, these enzymes aid in the "unwinding" of DNA so that it can be replicated during cell division. If they are bound by a quinolone antibiotic, they are prevented from performing this function. Resistance to quinolones is chromosomally mediated; plasmid-mediated resistance has not been described. The primary mechanisms used in quinolone resistance are alterations in the target enzymes or alterations in bacterial permeability to the antibiotic. Many bacteria use several resistance mechanisms simultaneously, which may explain the very rapid rate of the development of resistance to quinolones.54 In a 1991 report, hospital policy was changed to foster the use of quinolones to treat MRSA. When the policy was instituted, there was virtually no resistance to quinolones among the staphylococcus isolates. Within one year, quinolone resistance topped 70%.55 A similar outcome was documented in another study that examined the rate of quinolone resistance among Campylobacter isolates. Virtually all isolates from U.S. military personnel in Thailand in 1990 were susceptible to ciprofloxacin. By 1994, 69-79% of isolates were resistant. This rapid rise in resistance was probably due to two factors: the common use of quinolones in the empiric treatment of traveler’s diarrhea, and the widespread use of antibiotics in poultry and other livestock.56,57
Current Concerns
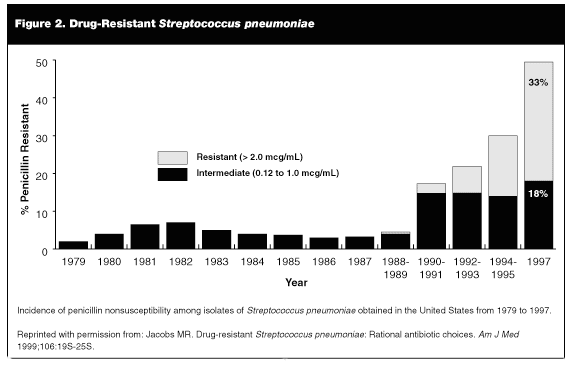
Penicillin-Resistant Pneumococcus (PRP). The first isolate of PRP was described in 1967 in Papua, New Guinea. Isolates are classified as sensitive, intermediate, or resistant on the basis of the concentration of penicillin required to inhibit growth. Currently, the cut points in use are: sensitive, minimal inhibitory concentration (MIC) less than 0.1 mcg/mL penicillin; intermediate, MIC between 0.1 and 1.0 mcg/mL; and resistant, MIC higher than 2.0 mcg/mL. Resistance appears to have been acquired from other organisms, since foreign DNA sequences have been identified in the resistance genes. It is common to find alterations in many different PBPs within the same organism, so a resistant strain actually may have many resistance genes.40
Resistance remained relatively rare until the late 1980s, when rates still hovered around 3-6%.58 A dramatic rise in levels of resistance occurred in the 1990s, with up to 44% of isolates showing either intermediate or high level resistance by 1997.59 Fortunately, increased resistance has not lead to increased virulence.60,61 A study in 1995 showed no increase in morbidity or mortality in children with pneumonia caused by PRP.62
There is a strong correlation between the widespread use of antibiotics and the development of PRP. Numerous studies, particularly in day care centers and schools, have shown that recent antibiotic use increases the risk of PRP colonization in children.63,64 Moreover, that risk appears to extend to the children who have not had recent antibiotic exposure, most likely due to clonal spread from children with PRP to children without it.65
Vancomycin-Resistant Enterococcus. Resistance to vancomycin among the enterococci was first reported in 1986.66 Van genes have thus far been detected only in enterococci, but resistance plasmids have been transferred in vitro to S. aureus, opening up the frightening prospect that staphylococcus could acquire the Van genes naturally.67 In vitro transfer of glycopeptide resistance plasmids also has been demonstrated for Listeria monocytogenes and streptococci.51 VRE rarely was isolated in U.S. hospitals before 1990, but by 1998 vancomycin resistance was found in more than 20% of all hospital isolates of enterococci.68 The rate of increase of VRE mirrors that of PRP, so much higher resistance can be expected with each passing year. Data have consistently shown that there is a strong correlation between the use of broad-spectrum antibiotics and the development of VRE, particularly antibiotic regimens aimed at gram-negative pathogens.69-70 Not surprisingly, the rates tend to be significantly higher in intensive care units than on the wards within the same hospital.71
The spread of VRE in this decade has lead to a crisis not unlike the one caused by penicillin-resistant staphylococcus in the 1950s. In many cases, people who developed life-threatening VRE infections were left with no available therapy, although various combinations of alternative antibiotics have been effective in some. Then, in the late 1990s, several antibiotics from completely new classes were developed. The first to be released was quinupristin/dalfopristin (Synercid), the first of a class of antibiotics known as streptogramins.72 Next came the release of linezolid from the new class of oxazolidonones.73 These antibiotics offer some hope for patients who are infected with VRE or MRSA, but already quinupristin/dalfopristin-resistant enterococci have been described.74 A particular concern is that the use of related compounds to promote growth in poultry may compromise the effectiveness of quinupristin/dalfopristin in humans.75
Methicillin-Resistant Staphylococcus aureus. Resistance quickly developed in S. aureus after the introduction of penicillin and then spread throughout the world in less than a decade. This resistance was accelerated by the clonal spread of plasmids carrying the resistance genes. Affected organisms produced inducible beta-lactamases, so that even very high doses of penicillin were unhelpful in these infections. By the end of the 1950s, there was virtually no available therapy for those patients who developed life-threatening infections due to S. aureus.13 Because of this, the development and release of methicillin in 1961 was hailed as a major advance. However, within less than a year, MRSA was reported. This time, resistance was mediated through alteration of the PBPs. The resistance gene is transposable, further facilitating its spread. The rise in resistance was particularly fast among isolates of Staphylococcus epidermidis, and less so among isolates of S. aureus. In 1975, the rate of isolation of MRSA in U.S. hospitals was still only 2.4%, but by 1991 it had increased to 29%.76 Throughout the 1990s, MRSA rates increased dramatically. By 1999, MRSA accounted for 53% of S. aureus infections in U.S. intensive care units.77
Most MRSA isolates are resistant to multiple antibiotics. No isolate of S. aureus has been reported to be fully resistant to vancomycin, although the first reports of intermediate level resistance were published in 1997.78 It appears that intermediate resistance to vancomycin occurs in a step-wise fashion when MRSA is exposed to vancomycin for prolonged periods. It’s presence can be difficult to detect by standard laboratory methods, so clinicians must be alert to its possible role in patients whose MRSA infections fail to respond to vancomycin. 79 The inevitability of fully vancomycin-resistant S. aureus already has prompted discussions about how the medical community should respond to its arrival.80 As is the case with VRE, MRSA rates vary from hospital to hospital, but the most dramatic differences correlate with the size of the hospital. In 1992, rates of MRSA in hospitals with more than 500 beds was twice that of hospitals with fewer than 200 beds. This is probably a reflection of increased complexity of illness and more broad spectrum antibiotic use at the larger, tertiary care hospitals.81
Multi-Drug Resistant Tuberculosis (MDR-TB). Perhaps no microorganism better exemplifies the link between inappropriate antibiotic use and the development of resistance than MTB. In this example, improper prescription of antimicrobials is not the cause of resistance. Rather, it is improper use of the prescribed antibiotics that leads to resistance. Incomplete, sporadic, or erratic compliance by infected individuals is a recipe for MDR-TB.
 |
Soon after therapy for TB became available 50 years ago, it quickly became evident that a single antibiotic was not likely to cure the patient with active TB, and in fact, resistant TB often resulted. Resistance to antituberculous drugs is chromosomally mediated; a background resistance rate of 10-5 to 10-7 can be expected with any of the major drugs used for TB therapy.42 This means that in a typical cavitary lesion, which might contain 109 organisms, one should expect to find 10 to 1000 organisms that are resistant to isoniazid (INH), rifampin, pyrazinamide, or ethambutol. This explains the failure of single-drug therapy for cavitary disease. Fortunately, resistance genes are not linked, so the likelihood that an isolate will be resistant to two drugs simultaneously is between 10-10 and 10-14. Thus, in the aforementioned cavitary lesion, the odds are heavily against an isolate that is naturally resistant to both INH and rifampin. These facts have lead to the current practice of providing multiple antituberculous drugs to patients with active disease.
MDR-TB refers to isolates that are resistant to at least INH and rifampin. In 1998, 1.1% of all MTB isolates tested in the United States showed multi-drug resistance.82 A common misconception is that the rise of MDR-TB during the 1990s was caused by the importation of a "superbug" from another part of the world. In fact, most MDR-TB in the United States is acquired, which is defined as resistance in a patient who previously has received antituberculosis medications. Acquired resistance is generally due to improper treatment or noncompliance. Primary resistance refers to MDR-TB that is resistant before any therapy has been given to the patient. Between 1994 and 1997, about 20% of multi-drug resistant isolates in the United States showed primary multi-drug resistance, while 80% was acquired.83 In theory, primary multi-drug resistance may be the result of random mutation, but spread of MDR-TB from an index patient to someone he or she comes into contact with is far more likely.
Consider a patient with active pulmonary TB. The patient is correctly diagnosed, and sputum cultures reveal MTB that is sensitive to all the major antituberculosis medications. The patient is appropriately placed on a regimen of INH, rifampin, and pyrazinamide. He fills the prescriptions for INH and rifampin alone. He takes his combination therapy for one or two doses, then realizes that the rifampin is turning his urine red. He decides to stop taking rifampin. Of the 108 organisms in his cavitary lesion, 106 respond quickly to the INH, and the number of viable organisms diminishes rapidly. The patient begins to feel better. However, 102 of the original organisms are resistant to INH, and are unaffected by therapy. Soon, the only organisms left in the cavity are those that are resistant to INH, and they begin to multiply in the less crowded environment. In several more weeks, the cavity is occupied by 108 organisms once again, except that now they are 100% resistant to INH. At this point, the patient begins to feel ill once more, so he begins to take the rifampin as initially directed. 107 organisms respond to the rifampin and soon die, but 101 organisms are resistant to rifampin. These organisms then expand their numbers in the cavity so that, in several weeks, 108 organisms are present once more, all of which are 100% resistant to both INH and rifampin. A case of MDR-TB has just been created.
Strategies for Battling Resistance
It may seem that we humans are hopelessly outmatched by the microbes in our struggle for survival. It is true that we can never achieve full control over bacteria and the infections they cause with our current approach, but we do have some tools at our disposal to aid the effort. Wise use of these tools can result in countless lives saved or improved.
Public Health Efforts. While much of the effort to combat antibiotic resistance must come from individual practitioners making individual decisions every day, public health programs are uniquely suited to address some of these problems. An obvious example is the strategy now being employed by cities across the United States to combat MDR-TB. The homeless population, those with low incomes, inner-city residents, and drug addicts are among the highest risk groups for infection by MTB. These groups also are those most at risk for non-compliance with medications. If TB were an acute illness such as gram-negative sepsis that runs its course over a short time period, this danger would be confined largely to the individual with the disease. But active TB tends to run a much more indolent course, with some patients surviving for months or even years despite active infection. During this time both the disease itself and drug resistance can be disseminated throughout the community. Thus, the public has a great stake in seeing that TB is treated properly.
Largely on the strength of this argument, many cities in the United States have empowered their public health officials to direct therapy for TB. One of the most successful strategies used has been directly observed therapy (DOT), whereby a public health worker supervises the administration of the oral medication, usually at the public health clinic. A study from Texas in 1994 illustrates the value of this strategy. From 1980 to 1986, therapy was prescribed for active TB but was unobserved. During this period, the rated of acquired resistance rose to 14%, and the relapse rate was 20.9%. Both of these outcomes would be expected with poorly compliant patients. From 1986 to 1992, directly observed therapy was used for all new cases of active TB. By 1992, the rate of acquired resistance dropped to 2%, and the relapse rate dropped to 5.5%.84 This approach has been so successful that some cities even provide for temporary incarceration in a hospital setting for those who refuse to comply. By the end of the 1990s, rates of TB and multi-drug resistance were substantially lower than they were 10 years earlier, a decline that appears to closely correlate with TB control measures.82
There also are major collaborative efforts between professional organizations and public health authorities to spread the word about inappropriate antibiotic use to the general public. For example, the American Academy of Pediatrics, Centers for Disease Control and Prevention, and The American Society for Microbiology have collaborated to produce a brochure entitled "Your Child and Antibiotics: Unnecessary Antibiotics CAN Be Harmful." Copies can be obtained through American Academy of Pediatrics, which is based in Elk Grove Village, IL. Further work is needed to evaluate the effect of these interventions.
Restrict Extended-Spectrum Antibiotics. Many hospitals have policies in place to restrict the use of extended spectrum antibiotics, including cephalosporins, monobactams, and vancomycin. The aim of these policies is to reduce the rate of increase of resistance by pressing physicians to use antibiotics with a narrower spectrum of activity. These policies have been extensively studied, with mixed results.5,85 In one study, an antibiotic restriction policy was begun in an effort to control cephalosporin resistance.45 During the study period, authorization was required to use extended spectrum cephalosporins. Exempted categories included children, single-use surgical prophylaxis, bacterial meningitis, bacterial peritonitis, and the outpatient treatment of gonococcal infections. During the study period there was an 80% reduction in cephalosporin use throughout the hospital, and a 44% reduction in ceftazidime-resistant Klebsiella infection. Another study at Ben Taub Hospital in Houston examined the effect of requiring prior authorization for selected antimicrobials.86 Authorization was required for the use of amikacin, ciprofloxacin, fluconazole, ofloxacin, timentin, piptazocillin, and aztreonam. During the study period total expenditures for antimicrobials decreased by 32%, and susceptibilities to all beta-lactam and quinolone antibiotics increased. There were no adverse effects on patient care. In a 1996 study, gastrointestinal tract colonization with VRE was cut from 47% to 15% in just six months when vancomycin, clindamycin, and third-generation cephalosporin use was restricted.87 While these studies and others like them are promising, these policies require a concerted effort, and if the restrictions are loosened, a quick return of resistance can result. Studies of extended spectrum antibiotic restriction programs have not been conducted in the outpatient setting, but similar reductions in resistance could be expected.
Decrease Selective Pressure of Oral Antibiotics. The widespread use of antibiotics has created a selective pressure against sensitive organisms and in favor of resistant organisms. Will a decrease in the use of these antibiotics reverse the trend? Perhaps. Studies examining the cost of resistance suggest that, in the absence of antibiotics, the carriage of resistance plasmids may place bacterial populations at a competitive disadvantage relative to non-plasmid containing bacteria.44 Other authors cite data showing stable inheritance of resistance plasmids even in the absence of antibiotic-mediated selection, and suggest a bleak outlook for control of resistance through antibiotic restriction programs.85,88 Some studies have shown a promising reduction in the rates of resistance with decreased antibiotic use.
A 1995 study examined the rate of penicillin-resistant S. pneumoniae in a day care center.65 Among the 59% of children who were colonized with S. pneumoniae in April, 53% had highly resistant isolates, and 22% had intermediately resistant isolates. Seventy-five precent of these children had completed a course of antibiotics in the preceding two months. Upon re-examination four months later, 54% were found to be colonized with S. pneumoniae, but only 7% of strains were highly resistant, and 39% were intermediately resistant. This time, only 46% had taken any antibiotics in the preceding two months. The authors concluded that the decrease in antibiotic use among the day care attendees was likely the cause for the lower PRP rates. Another important finding in this study was that children in the same day care classroom tended to share the same pulsed field gel electrophoresis strain types, indicating that horizontal spread of PRP had occurred among classmates.
A 1994 study from Japan looked at the rate of erythromycin resistance among group A streptococcus (GAS) isolates.89 In the early 1970s, erythromycin accounted for 22% of all the antibiotics used in the country, and by 1975, GAS showed a resistance rate to erythromycin of 62%. Soon after, a national campaign was undertaken to reduce the use of erythromycin. By 1988, erythromycin accounted for only 8% of all the antibiotics used. GAS resistance to erythromycin dropped to 22% in 1982, and has been virtually absent since 1986.
A study published in 1996 examined rates of antibiotic resistant organisms among children who were prescribed amoxicillin or sulfa for otitis media prophylaxis.90 These authors showed a dramatic increase in both beta-lactamase producing organisms and PRP while the children were on amoxicillin, and conversely, a dramatic decline in both after cessation of therapy.
Stop Unwarranted Antibiotic Use. The most obvious method to reduce antibiotic use and the spread of resistant organisms is to stop the common practice of prescribing antibiotics for non-bacterial illnesses. Gone are the days when a physician could justify the use of antibiotics for a patient with a cold by reasoning that "it can’t do any harm." This casual attitude has, in part, brought us to our current resistance problem. Antibiotics are effective against bacteria, and should be used for illnesses that are likely to be bacterial in origin. They are not effective against viruses, and are rarely indicated for illnesses that are likely to be viral in origin. Fever is a common symptom in the pediatric population, but serious bacterial infections account for only 2-9% of all fevers in children.91-92 Viral infections account for substantially more than one-half of all fevers in children, and otitis media and sinusitis account for most of the rest. Given the high rate of spontaneous resolution of otitis media and sinusitis, it can be concluded that fewer than 15% of children with fever truly need immediate antibiotics. And yet, more than one-half of pediatricians and family practitioners surveyed in a recent study prescribed antibiotics for the common cold.23 Studies such as this have led to major efforts by national organizations to curtail antibiotic use.
The American Academy of Pediatrics has been a particularly prominent advocate for a more rational use of antibiotics. In 1998, the organization joined with the Centers for Disease Control and Prevention to co-sponsor a supplement to Pediatrics titled "Principles of Judicious Use of Antimicrobial Agents for Pediatric Upper Respiratory Tract Infections."93 This publication provides a short but excellent review of the available literature on the natural history and treatment of otitis media, pharyngitis, acute sinusitis, cough illness/bronchitis, and the common cold. An update on these recommendations can be found in the September 2000 issue of Pediatrics.94 A common theme is present in all of the recommendations found in these articles. First, most of these respiratory illnesses, including otitis media and sinusitis, either have a viral etiology or can be expected to resolve without antibiotic therapy. Spontaneous resolution of symptoms can be expected in 80-85% of patients with acute otitis media and in more than 50% of patients with sinusitis. Some European countries now follow protocols whereby children with acute otitis media are treated with antibiotics only if their symptoms persist beyond two days.28,95 Second, antimicrobial therapy should be directed at the most likely bacterial pathogens. This requires that health care workers maintain an awareness of community resistance patterns and changing recommendations for treating bacterial infections such as acute otitis media.49 Third, we must be careful to use consistent criteria to diagnose infections such as acute otitis media and sinusitis. We do our patients no favors by over-diagnosing these conditions and prescribing antibiotics unnecessarily. (See Table 3.)
| Table 3. Antibiotic Guidelines93 |
| Pharyngitis |
| • Most cases are due to viruses. |
| • An exam alone is inadequate to establish the presence of group A streptococcus (GAS) infection. |
| • Rapid antigen detection test and/or culture should be done to confirm GAS. |
| • Treat only confirmed cases of GAS; empiric therapy is not warranted. |
| • Penicillin is the drug of choice. |
| Upper Respiratory Infection (Common Cold) |
| • Fever lasts 3-5 days, other symptoms 10-14 days. |
| • Colored/mucopurulent discharge is part of the natural history, and is not predictive for sinusitis. |
| • Antibiotics are warranted only for complications. |
| Cough Illness/Bronchitis |
| • Virtually always viral in origin in children. |
| • Fever is common during first 3-5 days. |
| • Cough commonly persists for up to 10 days. |
| • Antibiotics may be warranted in cases of prolonged fever or persistent cough. |
| Otitis Media |
| • Acute otitis media (AOM): Middle ear effusion with signs or symptoms of acute local or systemic illness. |
| • Otitis media with effusion (OME): Middle ear effusion in the absence of signs or symptoms of acute infection. |
| • Spontaneous cure rate is 80-85% in AOM. |
| • Red tympanic membrane alone is not diagnostic for AOM. |
| • OME should not be treated with antibiotics unless chronic. |
| • Half-life of fluid behind a tympanic membrane is 3-4 weeks. |
| Sinusitis |
| • Viral rhinosinusitis is 20-200 times more common than bacterial. |
| • Spontaneous cure rate is 60%. |
| • Treat for prolonged symptoms (> 10-14 days) or for severe symptoms. |
| • Radiographs generally not indicated or helpful. |
Conclusion
Good physicians are trained to use their judgment, based on the known science, when treating individual patients. This is what is meant by the art of medicine. Policies that may be good for the population as a whole may be seen by some to be detrimental to the individual patient. However, on the subject of antibiotics, one issue has become clear: When antibiotics are used inappropriately, not only does the individual patient fail to benefit, but adverse effects also can be expected both for the individual and for the community. Our arsenal is limited, and the bacteria quickly develop countermeasures for anything we use against them. Thus, it is incumbent upon all physicians to use antibiotics selectively and wisely. In this way we can delay our eventual defeat by the microbes. The wise use of antibiotics is good policy for individuals and for the community.
References
1. Cohen ML. Changing patterns of infectious disease. Nature 2000;406:
762-767.
2. Acar JF. Consequences of bacterial resistance to antibiotics in medical practice. Clin Infect Dis 1997;24(Suppl 1):S17-S18.
3. Schwartz B, Bell DM, Hughes JM. Preventing the emergence of antimicrobial resistance: A call for action by clinicians, public health officials, and patients. JAMA 1997;278:944-945.
4. Kunin CM. Editorial response: Antibiotic armageddon. Clin Infect Dis 1997;25:240-241.
5. Burke JP. Antibiotic resistance: Squeezing the balloon? JAMA 1998;280:
1270-1271.
6. Gold HS, Moellering RC. Antimicrobial drug resistance. N Engl J Med 1996;335:1445-1453.
7. Holmberg SD, Solomon SL, Blake PA. Health and economic impacts of antimicrobial resistance. Rev Infect Dis 1987;9:1065-1078.
8. Scientific Working Group on Antibiotic Resistance. Clin Infect Dis 1997;24(Suppl 1):S1-S176.
9. Hughes VM, Datta N. Conjugative plasmids in bacteria of the "pre-antibiotic" era. Nature 1983;302:725-726.
10. Rammelkamp CH, Maxon T. Resistance of Staphylococcus aureus to the action of penicillin. Proc Soc Exp Biol Med 1942;51:386-389.
11. Jevons MP. Celbenin-resistant staphylococci. BMJ 1961;1:124-125.
12. Datta N, Kontomichalou P. Penicillinase synthesis controlled by infectious R factors in enterobacteriaceae. Nature 1965;208:239-241.
13. Medeiros AA. Evolution and dissemination of beta-lactamases accelerated by generations of beta-lactam antibiotics. Clin Infect Dis 1997;24(Suppl):
S19-S45.
14. Hart CA, Percival A. Resistance to cephalosporins among gentamicin-resistant Klebsiellae. J Antimicrob Chemother 1982;9:275-286.
15. Knothe H, Shah P, Kremery V, et al. Transferable resistance to cefotaxime, cefoxitin, cefamandole and cefuroxime in clinical isolates of Klebsiella pneumoniae and Serratia marcescens. Infection 1983;11:315-317.
16. Yang Y, Wu P, Livermore DM. Biochemical characterization of a beta-lactamase that hydrolyzes penems and carbapenems from two Serratia marcescens isolates. Antimicrob Agents Chemother 1990;34:755-758.
17. O’Brien TF. The global epidemic nature of antimicrobial resistance and the need to monitor and manage it locally. Clin Infect Dis 1997;24(Suppl):S2-S8.
18. Van der Auwera, Pensart N, Korten V, et al. Influence of oral glycopeptides on the fecal flora of human volunteers: Selection of highly glycopeptide-resistant enterococci. J Infect Dis 1996;173:1129-1136.
19. Macris MH, Hartman N, Murray B, et al. Studies of the continuing susceptibility of group A streptococcal strains to penicillin during eight decades. Pediatr Infect Dis J 1998;17:377-381.
20. Hoiby N, Jarlov JO, Kemp M, et al. Excretion of ciprofloxacin in sweat and multiresistant Staphylococcus epidermidis. Lancet 1997;349:167-169.
21. Mainous AG, Hueston WJ, Clark JR. Antibiotics and upper respiratory infection: Do some folks think there is a cure for the common cold? J Fam Pract 1996;42:357-361.
22. Gonzales R, Steiner JF, Sande MA. Antibiotic prescribing for adults with colds, upper respiratory tract infections, and bronchitis by ambulatory care physicians. JAMA 1997;278:901-904.
23. Schwartz RH, Freij BJ, Ziai M, et al. Antimicrobial prescribing for acute purulent rhinitis in children: A survey of pediatricians and family practitioners. Pediatr Infect Dis J 1997;16:185-190.
24. Davy T, Dick PT, Munk P. Self-reported prescribing of antibiotics for children with undifferentiated acute respiratory tract infections with cough. Pediatr Infect Dis J 1998;17:457-462.
25. Rosenstein N, Phillips WR, Gerber MA, et al. The common cold—Principles of judicious use of antimicrobial agents. Pediatrics 1998;101:181-184.
26. Jacobs RF. Judicious use of antibiotics for common pediatric respiratory infections. Pediatr Infect Dis J 2000;19:938-943.
27. Gadomski AM. Potential interventions for preventing pneumonia among young children: Lack of effect of antibiotic treatment for upper respiratory infections. Pediatr Infect Dis J 1993;12:115-120.
28. Dowell SF, Marcy SM, Phillips WR, et al. Otitis media—principles of judicious use of antimicrobial agents. Pediatrics 1998;101:181-184.
29. Paradise JL. Managing otitis media: A time for change. Pediatrics 1995;96:
712-715.
30. Gleckman RA, Borrego F. Adverse reactions to antibiotics. Postgraduate Medicine 1997;101:97-108.
31. Dowell SF, Schwartz B. Resistant pneumococci: Protecting patients through judicious use of antibiotics. Am Fam Physician 1997;55:1647-1654.
32. Boken DJ, Chartrand SA, Goering RV, et al. Colonization with penicillin-resistant Streptococcus pneumoniae in a child-care center. Pediatr Infect Dis J 1995;14:879-884.
33. Wong CS, Jelacic S, Habeeb RL, et al. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Eschericia coli 0157:H7 infections.
N Engl J Med 2000;342:1930-1936.
34. Droste JHJ, Wieringa MH, Weyler JJ, et al. Does the use of antibiotics in early childhood increase the risk of asthma and allergic disease? Clin Exp Allergy 2000;30:1547-1553.
35. Mattes J, Karmaus W. The use of antibiotics in the first year of life and development of asthma: Which comes first? Clin Exp Allergy 1999;29:729-732.
36. Bauchner H, Pelton SI, Klein JO. Parents, physicians, and antibiotic use. Pediatrics 1999;103:395-398.
37. Hamm RM, Hicks RJ, Bemben DA. Antibiotics and respiratory infections: Are patients more satisfied when expectations are met? J Fam Pract 1996;43:56-62.
38. Cowan P. Patient satisfaction with an office visit for the common cold.
J Fam Pract 1987;24:412-413.
39. Medeiros AA. Evolution and dissemination of beta-lactamases accelerated by generations of beta-lactam antibiotics. Clin Inf Dis 1997;24(Suppl 1):S19-S45.
40. Tomasz A. Antibiotic resistance in Streptococcus pneumoniae. Clin Inf Dis 1997;24(Suppl 1):S85-S88.
41. Miller GH, Sabatelli FJ, Hare RS, et al. The most frequent aminoglycoside resistance mechanisms—Changes with time and geographic area: A reflection of aminoglycoside usage patterns? Clin Inf Dis 1997;24(Suppl):S46-S62.
42. Zgurskaya HI, Nikaido H. Multi-drug resistance mechanisms: Drug efflux across two membranes. Molecular Microbiology 2000;37:219-225.
43. Jacobs RF. Multiple-drug-resistant tuberculosis. Clin Inf Dis 1994;19:1-10.
44. Hayden MK. Insights into the epidemiology and control of infection with vancomycin-resistant enterococci. Clin Inf Dis 2000;31:1058-1065.
45. Levin BR, Lipstitch M, Perrot V, et al. The population genetics of antibiotic resistance. Clin Inf Dis 1997;24(Suppl 1):S9-S16.
46. Rahal JJ, Urban C, Horn D, et al. Class restriction of cephalosporin use to control total cephalosporin resistance in nosocomial Klebsiella. JAMA 1998;280:1233-1237.
47. Boyce JM, Jackson MM, Pugliese G. Methicillin-resistant Staphylococcus aureus (MRSA): A briefing for acute care hospitals and nursing facilities. Infect Control Hosp Epidemiol 1994;15:105-115.
48. Pichichero ME, Casey JR, Mayes T, et al. Penicillin failure in streptococcal tonsillophyaryngitis: Causes and remedies. Pediatr Infect Dis J 2000;19:
917-923.
49. Wiener J, Quinn JP, Bradford PA, et al. Multiple antibiotic-resistant Klebsiella and Escherichia coli in nursing homes. JAMA 1999;281:517-523.
50. Dowell SF, Butler JC, Giebink GS, et al. Acute otitis media: Management and surveillance in an era of pneumococcal resistance—A report from the Drug-resistant Streptococcus pneumoniae Therapeutic Working Group. Pediatr Infect Dis J 1999;18:1-9.
51. Bush K, Tanaka SK, Bonner DP, et al. Resistance caused by decreased penetration of beta-lactam antibiotics into Enterobacter cloacae. Antimicrob Agents Chemother 1985;27:555-560.
52. Leclercq R. Enterococci acquire new kinds of resistance. Clin Inf Dis 1997;
24(Suppl):S80-S84.
53. Arthur M, Courvalin P. Genetics and mechanisms of glycopeptide resistance in Enterococci. Antimicrob Agents Chemother 1993;37:1563-1571.
54. Schmitz FJ, Fluit AC, Hafner D, et al. Development of resistance to ciprofloxacin, rifampin, and mupirocin in methicillin-susceptible and resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 2000;
44:3229-3231.
55. Blumberg HM, Rimland D, Carroll DJ, et al. Rapid development of ciprofloxacin resistance in methicillin-susceptible and -resistant Staphylococcus aureus. J Infect Dis 1991;163:1279-1285.
56. Murphy GS Jr, Echeverria P, Jackson LR, et al. Ciprofloxacin- and azithromycin-resistant Campylobacter causing traveler’s diarrhea in U.S. troops deployed to Thailand in 1994. J Infect Dis 1996;22:848-869.
57. Ferber D. Superbugs on the hoof? Science 2000;288:792-794.
58. Doern GV, Brueggemann A, Holley HP, et al. Antimicrobial resistance of Streptococcus pneumoniae recovered from outpatients in the U.S. during the winter months of 1994 to 1995: Results of a 30-center national surveillance study. Antimicrob Agents Chemother 1996;40:1208-1213.
59. Doern GV, Pfaller MA, Kugler K, et al. Prevalence of antimicrobial resistance among respiratory tract isolates of Streptococcus pneumoniae in North America: 1997. Results from the SENTRY Antimicrobial Surveillance Program. Clin Inf Dis 1998;27:764-770.
60. Silverstein M, Bachur R, Harper MB. Clinical implications of penicillin and ceftriaxone resistance among children with pneumococcal bacteremia. Pediatr Infect Dis J 1999;18:35-41.
61. Friedland I, McCracken G. Management of infections caused by antibiotic resistant Streptococcus pneumoniae. N Engl J Med 1994;331:377-382.
62. Friedland IR. Comparison of the response to antimicrobial therapy of penicillin-resistant and penicillin-susceptible pneumococcal disease. Pediatr Infect Dis J 1995;14:885-890.
63. Jacobs MR. Drug-resistant Streptococcus pneumoniae: Rational antibiotic choices. Am J Med 1999;106:S19-S25.
64. Reichler MR, Allphin AA, Breiman RF, et al. The spread of multiply resistant Streptococcus pneumoniae at a day care center in Ohio. J Infect Dis 1992;166:1346-1353.
65. Nava JM, Bella F, Garau J, et al. Predictive factors for invasive disease due to penicillin-resistant Streptococcus pneumoniae: A population-based study. Clin Infect Dis 1994;19:884-890.
66. Leclercq R, Derlot E, Duval J, et al. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 1988;319:157-161.
67. Noble WC, Virani Z, Cree R. Cotransfer of vancomycin and other resistance genes from Enterococcus fecalis NCTC 12201 to Staphylococcus aureus. FEMS Microbiol Lett 1992;93:195.
68. Fridkin SK, Gaynes RP. Antimicrobial resistance in intensive care units. Clin Chest Med 1999;20;303-316.
69. Hayden MK. Insights into the epidemiology and control of infection with vancomycin-resistant enterococci. Clin Infect Dis 2000;31:1058-1065.
70. Boyce JM. Vancomycin-resistant enterococcus: Detection, epidemiology, and control measures. Infect Dis Clin North Am 1997;11:367-384.
71. Archibald L, Phillips L, Monnet D, et al. Antimicrobial resistance in isolates from inpatients and outpatients in the U.S.: Increasing importance of the intensive care unit. Clin Infect Dis 1997;24:211-215.
72. Synercid. Medical Letter 1999;41:109.
73. Linezolid (Zyvox). Medical Letter 2000;42:45-46.
74. Chow JW, Donahedian SM, Zervos MJ, et al. Emergence of increased resistance to quinupristin/dalfopristin during therapy for Enterococcus faecium bacteremia. Clin Infect Dis 1997;24:91-92.
75. Angulo F, Marano N, MacKinson C, et al. Isolation of quinupristin/dalfopristin-resistant Enterococcus faecium from human stool specimens and retail chicken products in the U.S.—Associated with use of virginiamycin in poultry? First International Conference on Enterococci. Banff, Canada; February 2000.
76. Panlilio AL, Culver DH, Gaynes RP, et al. Methicillin-resistant Staphylococcus aureus in U.S. hospitals, 1975-1991. Infect Control Hosp Epidemiol 1992;13:582-586.
77. National Nosocomial Infections Surveillance (NNIS) System report, data summary from Jan 1992-April 2000. Am J Infect Control 2000;28:429-448.
78. Martin R, Wilcox KR, Campbell C, et al. Update: Staphylococcus aureus with reduced susceptibility to vancomycin United States 1997. MMWR Morb Mortal Wkly Rep 1997;46:813-815.
79. Waldvogel FA. New resistance in Staphylococcus aureus. N Engl J Med 1999;340:556-557.
80. Edmond MB, et al. Vancomycin-resistant Staphylococcus aureus: Prospective measures needed for control. Ann Intern Med 1996;124:329-334.
81. Cormican MG, Jones RN. Emerging resistance to antimicrobial agents in gram-positive bacteria: Enterococci, staphylococci and nonpneumococcal streptococci. Drugs 1996;51(Suppl )1:6-12.
82. Progress toward the elimination of tuberculosis. Morb Mortal Wkly Rep 1999;48:732-735.
83. Pablos-Mendez A, Raviglione MC, Laszlo A, et al. Global surveillance for antituberculosis-drug resistance 1994-1997. N Engl J Med 1998;338:
1641-1619.
84. Weis SE, Slocum PC, Blais FX, et al. The effect of directly observed therapy on the rates of drug resistance and relapse in tuberculosis. N Engl J Med 1994;330:1179-1184.
85. Fierer J, Guiney D. Extended-spectrum beta-lactamases: A plague of plasmids. JAMA 1999;281:563-564.
86. White AC, Atmar RL, Wilson J, et al. Effects of requiring prior authorization for selected antimicrobials: Expenditures, susceptibilities, and clinical outcomes. Clin Inf Dis 1997;25:230-239.
87. Quale J, Landman D, Saurina G, et al. Manipulation of a hospital-antimicrobial formulary to control an outbreak of vancomycin-resistant enterococci. Clin Infect Dis 1996;23:1020-1025.
88. Salyers AA, Amabile-Cuevas CF. Why are antibiotic resistance genes so resistant to elimination? Antimicrob Agents Chemother 1997;41:2321-2325.
89. Fujita K, Murono K, Yoshikawa M, et al. Decline of erythromycin resistance of Group A streptococci in Japan. Ped Infect Dis J 1994;13:1075-1078.
90. Brook I, Gober AE. Prophylaxis with amoxicillin or sulfisoxazole for otitis media: Effect on the recovery of penicillin-resistant bacteria from children. Clin Inf Dis 1996;22:143-145.
91. McCarthy PL. Acute infectious illness in children. Compr Ther 1988;14:51-57.
92. McCarthy PL, Sznadjderman S, Forsyth BC, et al. Mother’s clinical judgement: A randomized trial of the acute illness observation scales. J Pediatr 1990;116:200-206.
93. Dowell SF, Marcy SM, Phillips WR, et al. Principles of judicious use of antimicrobial agents for pediatric upper respiratory tract infections. Pediatrics 1998;101(Suppl):163-184.
94. Pichichero ME. Judicious use of antibiotics in pediatric respiratory infections 2000. Pediatr Infect Dis J 2000;19:907-943.
95. Little P, Gould C, Williamson I, et al. Pragmatic randomised controlled trial of two prescribing strategies for childhood acute otitis media. British Med J 2001;322:336-342.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.