Special Supplement: Acute Coronary Syndromes (ACS): Outcome-Optimizing Treatment Guidelines for Patients With and Without Procedural Coronary Intervention (PCI)
Acute Coronary Syndromes (ACS): Outcome-Optimizing Treatment Guidelines for Patients With and Without Procedural Coronary Intervention (PCI)
Part II: Evidence-Based Analysis of Antithrombin Therapy—Standard Heparin vs. Low Molecular Weight Heparins
Author: Kurt Kleinschmidt, MD, FACEP, Assistant Professor, University of Texas Southwestern Medical Center, Dallas; Associate Director, Department of Emergency Medicine, Parkland Memorial Hospital, Dallas, Tex.
Peer Reviewer: William J. Brady, MD, Associate Professor, Program Director, Department of Emergency Medicine, University of Virginia, Charlottesville, Va.
Attention SubscribersThis acute coronary syndromes supplement was originally published in Emergency Medicine Reports, November 20, 2000, volume 21, number 24. Emergency Medicine Reports is edited by Gideon Bosker, MD, FACEP, Special Clinical Projects and Medical Education Resources; Assistant Clinical Professor, Section of Emergency Services, Yale University School of Medicine; Associate Clinical Professor, Oregon Health Sciences University. |
Editor’s note—Coronary heart disease (CHD) remains the principal cause of mortality in the United States. Among acute coronary syndromes (ACS), acute myocardial infarction (AMI) is the leading single cause of death, with more than 1.5 million cases and more than 500,000 associated deaths per year. Of the more than 95 million annual visits to the emergency department in the United States, nearly 8 million (8.4%) are due to chest pain. Not all of these patients, however, suffer from AMI; in fact, approximately 3 million of these individuals will have a noncardiac diagnosis. Of the 5 million patients with a probable cardiac etiology, 20% will have an AMI, 16% will have unstable angina (UA), and 6% will die suddenly from a variety of causes.1
In part one of this three-part series, Kleinschmidt examined the role of antiplatelet agents (e.g., aspirin, adenosine diphosphate [ADP] receptor antagonists, and glycoprotein IIb/IIIa inhibitors) in the management of patients with ACS, both with and without procedural coronary intervention (PCI). Relative advantages and disadvantages of specific agents within each class were discussed, and a strategy for sequenced, targeted use of antiplatelet drugs within the treatment pathway for ACS was outlined in detail.
In this issue, the principal focus is on antithrombin agents, direct thrombin inhibitors, standard (unfractionated) heparin (UFH), and low molecular weight heparins (LMWHs). Although both types of heparin are currently used for managing ACS, there is mounting evidence that superior outcomes can be achieved with the use of certain LMWHs in UA patients. In addition, it should be stressed that UFH has several disadvantages as an antithrombotic agent. At therapeutic levels, UFH can lead to thrombin formation by activating platelets. Also, thrombin generation has been reported after discontinuation of UFH.
UFH is difficult to administer, requiring continuous intravenous infusions and frequent monitoring of activated partial thromboplastin (aPTT). Finally, the incidence of heparin-induced thrombocytopenia (HIT) is significant, and is more common in patients receiving GP IIb/IIIa antiplatelet receptor inhibitors.
To circumvent the limitations and pitfalls of UFH—and also to evaluate the possibility of improving patient outcomes in ACS—LMWHs such as enoxaparin have been intensively studied as an evidence-based replacement for UFH in patients with UA and other acute coronary ischemic syndromes, including Non-ST Elevation Myocardial Infarction (NSTEMI).
From a pathophysiological perspective, LMWHs are more potent inhibitors of thrombin generation than UFH, and they are resistant to inhibition by activated platelets. Other benefits associated with LMWHs as compared to UFH include relatively simple dosing, ease of administration, limited requirements for further blood monitoring, and a more predictable anticoagulant effect.2-4 Finally, the rationale for use of LMWHs in acute coronary ischemic syndromes is supported by a number of evidentiary trials.
With these clinical issues in focus, the purpose of this issue is to examine the role of antithrombin agents for the management of ACS, and to develop an outcome-based strategy for using LMWHs and UFH in risk-stratified subgroups of patients with ACS. —Gideon Bosker, MD
Antithrombin Agents: Centerpiece Drugs for Management of Acute Coronary Syndromes
Overview: Antithrombin agents are a mainstay of therapy for patients with ACS. A number of antithrombin agents are available, and typically are differentiated according to their dependence upon antithrombin (AT) III. Both standard UFH and LMWHs depend upon AT III for their activity and are referred to as indirect thrombin inhibitors. Conversely, AT III-independent agents inhibit thrombin directly and do not require AT III for their activity.
Direct Thrombin Inhibitors: Hirudin is the most potent, naturally occurring, specific inhibitor of thrombin. Natural hirudin (from leech saliva), recombinant hirudin, and hirulog, a synthetic analog, are direct thrombin inhibitors. Each of these molecules binds directly and reversibly to thrombin at a 1:1 ratio both at the active site and to the fibrinopeptide-binding region of thrombin. Hirudin’s plasma half-life is 40 minutes following intravenous administration and approximately 120 minutes after subcutaneous injection.2 Hirulog has a plasma half-life of 24 minutes after intravenous infusion.5 Hirudin is approved for management of patients with heparin-induced thrombocytopenia and for ongoing anticoagulant therapy; however, it has not received approval for an ACS indication. In the setting of ACS, hirudin has been evaluated in patients with UA and NSTEMI; hirulog has been assessed primarily in patients undergoing percutaneous transluminal coronary angioplasty (PTCA).
Direct thrombin inhibitors have some theoretical advantages over indirect-acting heparins. These agents inhibit thrombin activity without affecting AT III, and they also suppress positive feedback mechanisms that promote further thrombin generation; in addition, they are not inactivated by platelet factor 4.6,7 These agents inactivate not only circulating thrombin but, unlike heparin, they also inhibit fibrin (clot)-bound thrombin. Because direct thrombin inhibitors do not bind endothelial cells and other plasma proteins, as does standard heparin, they have a more consistent dose-response and yield more predictable aPTTs.6,8 Unlike LMWHs, their use requires monitoring of the aPTT.7
Two studies have evaluated and compared clinical outcomes of hirudin vs. heparin in patients with ACS not undergoing PCI. They are: 1) the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO) IIb;9 and 2) the Organization to Assess Strategies for Ischemic Syndromes (OASIS) II trials.10 The GUSTO IIb trial evaluated 12,142 patients with an ACS. There was no significant difference in the primary end point of death or AMI at 30 days between patients treated with heparin or hirudin who did not have ST-segment elevation (9.1% vs 8.3%; P = 0.22).9 The OASIS II study included 10,141 patients with UA or NSTEMI who were randomized to receive hirudin (0.4 mg/kg bolus, followed by a 0.15 mg/kg/h infusion) or heparin (5000 U bolus followed by a 15 U/kg/h infusion). Drug dosages were adjusted to maintain a therapeutic aPTT. At 72 hours and at seven days there was a non-statistically significant trend toward less cardiovascular death or new AMI in the hirudin group.10
Direct thrombin inhibitors have been compared with heparin during angioplasty in the Hirulog Angioplasty Trial and the Hirudin Trial (HELVETICA).11,12 The Hirulog Angioplasty Trial was a randomized, double-blind comparison of heparin (175 U/kg bolus followed by an 18- to 24-hour infusion at 15 U/kg/h) with hirulog (1.0 mg/kg bolus followed by a 4-hour infusion at 2.5 mg/kg/h and then a 14- to 20-hour infusion at 0.2 mg/kg/h infusion).11 All patients received aspirin in both trials.
The incidence of the composite primary end point of in-hospital death, AMI, abrupt vessel closure, or rapid clinical deterioration of cardiac origin were comparable between the groups (11.4% for hirulog, 12.2% for heparin; P = 0.44). Major bleeding was significantly less frequent in those receiving hirulog compared with those receiving heparin (3.5% and 9.8%, respectively; P < 0.001).11 In the HELVETICA trial, hirudin therapy decreased the incidence of adverse clinical events at 96 hours after angioplasty vs. heparin therapy. However, there was no difference between the groups in the incidence of recurrent symptoms, clinical events, or restenosis at seven months.12
Standard Heparin (UFH)
Pharmacokinetics: UFH is a highly sulfated glycosaminoglycan found in mast cells of such animal tissues as lung, liver, and intestines. It is a mixture of polysaccharides with molecular weights between 5000 and 30,000 Daltons (average, 12,000-15,000). It has been the mainstay of anticoagulant therapy, and its clinical uses include prophylaxis against venous thromboembolism (VTE), acute management of thromboembolic disease, and as an integral component of multi-modal therapy for ACS.
Unfortunately, UFH has several shortcomings. Its use for acute thromboembolic disease or ACS requires admission to the hospital for intravenous administration. In addition, the pharmacokinetic properties of heparin make it difficult to achieve and maintain therapeutic levels; this may result in inadequate anticoagulation or over-anticoagulation with hemorrhagic complications. Heparin is neutralized by platelet factor 4, large quantities of which are released from platelets activated at sites of plaque rupture.
It should be stated that heparin does not inactivate activated factor X (factor Xa), which is bound to activated platelets trapped within the thrombus. From a thrombosis perspective, factor Xa activates prothrombin, and the resulting thrombin then binds to fibrin, where it also is protected from inactivation by heparin. As a result, fibrin- or clot-bound thrombin remains enzymatically active and accelerates thrombus growth through activation of local platelets and amplification of the coagulation system.2 As would be expected, frequent laboratory monitoring of the aPTT time is necessary to avoid these potential problems, which may contribute to increased risks for patients and increased cost of care.
Unstable Angina and Non-ST Elevation Myocardial Infarction. Heparin has been a mainstay of therapy for UA, NSTEMI, and ST-segment elevation AMI. Its benefit has been demonstrated in various trials comparing it to, and evaluating its efficacy in combination with, aspirin therapy.3,13,14 One study showed that heparin was superior to aspirin therapy in reducing cardiac events, particularly refractory angina, whereas others suggested it was at least as good as aspirin therapy. However, with heparin therapy alone, one study found rebound UA within the first 12 hours of cessation of heparin administration.15 This rebound phenomenon, which also was seen with other antithrombin agents, could be prevented by pretreatment with aspirin.
Based on these findings, it is now routine to treat patients early with a combination of aspirin and heparin—or LMWH—unless there is a contraindication. One double-blinded, randomized, placebo-controlled study compared aspirin, heparin, and aspirin plus heparin in 479 patients.3 Major end points included recurrent angina, MI, or death. All three arms had better outcomes than placebo. Patients receiving heparin had twice the bleeding compared to those treated with aspirin. Outcomes were similar among the three arms, with a trend toward heparin being better than aspirin alone.3
UFH has become a standard part of the management of UA. A number of randomized trials have suggested that heparin adds therapeutic benefit to aspirin in UA.3,13,16-19 However, the evidence is still not conclusive. The trials were small, only two were double-blind, and the confidence intervals were large. As is the case with GP IIb/IIIa trials, the populations in these heparin trials were sicker than many acute chest pain patients admitted to the hospital through the emergency department.
One group performed a meta-analysis of six small, UA studies that randomized patients to aspirin or a combination of aspirin and heparin. Pooled data from 1353 patients found a 33% reduction in progression to AMI or death with the addition of heparin, but this was of marginal statistical significance (P = 0.06).20 In these trials, heparin was associated with more major bleeding events, such as intracranial hemorrhage, requiring transfusion. The risk/benefit ratio must be considered before heparin is used in UA patients. In today’s environment, heparin should be used in moderate- or high-risk patients. Data do not support the routine use of heparin in low-risk patients.
Low Molecular Weight Heparin—The New Standard for ACS
In the late 1970s, it was recognized that it might be possible to dissociate the beneficial antithrombotic effects of heparin from its hemorrhagic anticoagulant effects. This insight provided the impetus to fractionate heparin and isolate the antithrombotic effects in the form of LMWHs. Compared with UFH, LMWHs have superior absorption and pharmacokinetic profiles, similar antithrombotic activities, and potentially fewer hemorrhagic complications. (See Table 1 by clicking here.) Moreover, LMWHs are proving to be at least as effective as heparin in a number of clinical settings, and they are revolutionizing the management of acute deep venous thrombosis by permitting home-based therapy.
Pharmacokinetics and Mechanisms of Action. LMWHs are fractions or fragments of heparin with molecular weights between 4000 and 6500 Daltons. All are produced by the fractionation of heparin molecules by controlled chemical or enzymatic depolymerization. Various depolymerization methods are used, resulting in many commercial LMWHs. It is important to recognize that each LMWH preparation has a distinct mean molecular weight, pharmacokinetic spectrum, and pharmacologic activity. The clinical relevance of these differences is not entirely clear, and randomized trials comparing the different LMWH preparations have yet to be done. However, regulatory authorities consider each LMWH preparation to be a distinct molecule, requiring its own documentation and FDA approval for specific clinical indications. Accordingly, the efficacy and safety features of one LMWH cannot be extrapolated to another, and each agent should be used according to evidence-based support and approved indications.
A number of LMWHs have been developed and approved for human use over the past decade. Interestingly, clinical use of LMWHs in the United States is relatively new, with four agents having received FDA approval: ardeparin (Normiflo), dalteparin (Fragmin), tinzaparin (Innohep), and enoxaparin (Lovenox). All four agents have been approved for management (i.e., prophylaxis and/or treatment) of VTE. Enoxaparin is the only LMWH approved for prophylaxis, inpatient and outpatient treatment of VTE, and acute management of ACS.4
Some generalizations can be made about the pharmacokinetics and metabolism of the different LMWHs. (See Table 1.) They are readily absorbed from the subcutaneous tissue, they are rapidly distributed to most organs and tissues, and they attain antithrombotic levels within 30 minutes of administration. Of special importance is the fact that LMWHs have greater than 90% bioavailability vs. approximately 30% for heparin. This difference in bioavailability is primarily related to heparin’s increased binding to plasma proteins, macrophages, and endothelial cells. Once bound, heparin’s antithrombotic activity is decreased because it can’t interact with the coagulation cascade proteinases. In addition, heparin binding is inconsistent, resulting in unpredictable activity.
The plasma half-life of LMWHs is two to four times longer than that of heparin, a feature that permits only once- or twice-daily administration. Elimination of heparin involves a rapid, saturable hepatic phase and a slower, renal clearance phase. The hepatic phase is dose-dependent, resulting in inconsistent elimination. In contrast, LMWHs primarily undergo slower renal elimination, which results in a longer half-life and more consistent elimination. The relative lack of platelet reactivity compared to heparin may result in less platelet activation and/or aggregation during an ACS. (See Table 1.)
Laboratory Monitoring: Antithrombotic and anticoagulant effects are different concepts. The beneficial antithrombotic properties reflect a molecule’s ability to prevent formation of a new thrombus or propagation of an existing thrombus. While it would be desirable to measure antithrombotic activity, there is no simple test for assessing antithrombotic activity. The anticoagulant effect of a substance reflects its ability to inhibit hemostasis, which may result in excessive bleeding. Anticoagulation activity can be measured indirectly by the activated aPTT, the test used to monitor the effect of heparin therapy. Unfortunately, the variable responsiveness of thromboplastin reagents used in the aPTT test can cause inconsistent results despite equivalent degrees of anticoagulation. Similar problems occurred with the use of prothrombin time ratios for the monitoring of oral anticoagulant therapy. This problem was overcome by standardizing the thromboplastin reagents and adapting the international normalized ratio (INR) system of reporting. However, no similar standardization for aPTT reagents is available currently.
The inaccuracy of the aPTT test for determining the anticoagulant effect of heparin is a problem because heparin’s dose response is inconsistent. The inconsistent dose response results from heparin’s low bioavailability and from its irregular elimination. Maintenance of therapeutic levels of heparin is difficult. Conversely, LMWH’s high and consistent bioavailability and dose-independent clearance result in a predictable anticoagulant response; therefore, laboratory monitoring is not needed in most patients. This is fortunate because the degree of anticoagulation induced by LMWH generally is too small to be detectable by the aPTT test. Anticoagulation should be monitored in patients receiving LMWHs who have renal insufficiency or those at the extremes of weight. This monitoring is done by a special test for anti-Xa activity.
Complications: Hemorrhage is the main complication of LMWH therapy. Bruising at the site of subcutaneous administration may occur, but this does not require alterations in therapy. While LMWH resulted in less hemorrhage than heparin in some early clinical trials, recent studies have revealed comparable hemorrhage rates. Caution must be used before giving LMWHs to patients at risk of hemorrhage. Neuroaxial hematomas have been reported in patients with the concurrent use of enoxaparin and spinal/epidural anesthesia. However, many of these events occurred prior to initiation of guidelines to decrease events.
Concomitant administration of LMWHs and other agents that impair hemostasis should be avoided. If a patient develops major bleeding complications secondary to a LMWH, protamine sulfate provides some but not complete neutralization of the antithrombin effect. In vitro studies have reflected that neutralization of antithrombin activity of LMWHs is virtually 100%, while that for anti-Xa ranges from 30-60% when protamine was used on a milligram per milligram basis.21 Little clinical information exists about the relative efficacy of protamine for stopping LMWH-related hemorrhage. One case report found reversal to be poor in a patient with a subdural hemorrhage who was inadvertently given enoxaparin 1 mg/kg bid for a period of four days. However, the protamine doses were only 20 mg; he eventually received a total dose of 100 mg.22
Both heparin and LMWHs may result in a transient decrease in platelet counts early in the course of therapy. Programs using LMWHs typically assess platelet counts every 2-3 days during therapy.23 One prospective series found a mild decrease in platelets in 28% of the patients receiving either heparin or LMWHs. This mild thrombocytopenia resolved in 99% of the patients within three days despite continuation of heparin or LMWH therapy.24 If the platelet count decreases to less than 100,000 per cubic millimeter, LMWH therapy should be discontinued.4
Heparin-induced thrombocytopenia (HIT) is a more serious platelet-heparin interaction. This process is a progressive, immune-mediated phenomenon typically occurring after 7-10 days of heparin therapy. It paradoxically results in hyperthrombosis and is associated with significant morbidity and mortality. Venous or arterial thromboses may occur, resulting in myocardial infarction, pulmonary emboli, and cerebrovascular accidents. Heparin-induced thrombocytopenia is associated less commonly with LMWHs than with heparin.24,25
One large series of 665 patients found that 2.7% of heparin-treated patients developed heparin-induced thrombocytopenia, while no cases occurred in those treated with LMWHs. The general consensus is that severe thrombocytopenia is much more rare with LMWHs than with UFH. Some of the patients treated with LMWHs did develop antiplatelet antibodies, which reflects the potential for developing heparin-induced thrombocytopenia.25 Both heparin and LMWH are contraindicated in patients who have previously had heparin-induced thrombocytopenia.
Up to 5% of patients receiving heparin or LMWHs have small, asymptomatic elevations of aspartate and alanine aminotransferases that reverse upon discontinuation of drug therapy. No monitoring guidelines exist for these aminotransferases. LMWH has limited transfer through the placenta, and teratogenic effects have not been noted. LMWHs are listed as category B drugs in pregnancy. It is unknown if LMWHs are excreted within the breast milk, and caution should be exercised when treating nursing women.
Comparison of LMWH with Standard Heparin: The differences in action between heparin and LMWH are better understood if the coagulation cascade (see Figure 1, below) is first reviewed. The cascade is a sequential, proteolytic activation of coagulation factor precursors (zymogens) into their active enzyme forms. Factors V and VIII serve as cofactors in the cascade. Most coagulation occurs via the extrinsic pathway, which is started by the interaction of factor VIIa with tissue factor, a plasma membrane protein on many cells. Tissue injury exposes tissue factor to VIIa, leading to the conversion of zymogen X to its active Xa form.
|
Figure 1. The Coagulation Cascade |
|
|
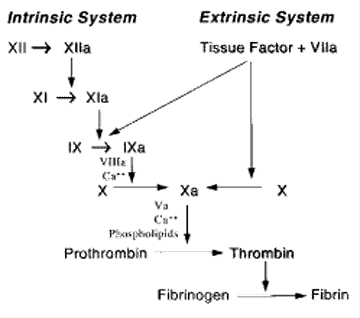
Tissue factor also activates zymogen IX, initiating the intrinsic pathway. Both the extrinsic and intrinsic pathways culminate in the activation of factor X and the formation of the prothrombinase complex on the platelet membrane. This prothrombinase complex contains calcium, phospholipids, active factor Xa, and cofactor Va. The complex converts prothrombin to thrombin, which converts fibrinogen to fibrin. While thrombin further supports thrombosis by activating platelets, it also counterbalances coagulation by facilitating the creation of antithrombin III and protein C, the principle anticoagulants.
Heparin’s anticoagulant activity requires antithrombin III. (See Figure 1.) Antithrombin III is the primary inhibitor of thrombin and the other proteinases IXa, Xa, XIa, and XIIa. Heparin binds to antithrombin III, producing a conformational change in the latter that potentiates its ability to rapidly inhibit the proteinases. Antithrombin III most easily suppresses thrombin (factor IIIa), while factor Xa is the most difficult factor for antithrombin III to inhibit because it is protected when bound within the prothrombinase complex.
The primary difference between heparin and LMWH is how they interact with thrombin. (See Figure 2.) Most heparin molecules are long; virtually all of these molecules are greater than 18 saccharide units in length. The long molecules can form a tertiary complex with antithrombin III and thrombin. When bound in this complex, thrombin is inactivated. Heparin molecules shorter than 18 saccharide units cannot form the tertiary complex and do not effectively inactivate thrombin. Because only 25-50% of LMWH molecules are more than 18 units in length, LMWH does not inactivate thrombin as well as does heparin. Conversely, factor Xa needs only a very short segment of heparin plus antithrombin III to be inactivated and it is equally inactivated by LMWH or by heparin. Thus, the anti-Xa to antithrombin inhibition ratio for heparin is 1:1, while it is 2-4:1 for LMWHs.
|
Figure 2. Interactions of Heparin and LMWH with Clotting Factors Xa and Thrombin |
|
|
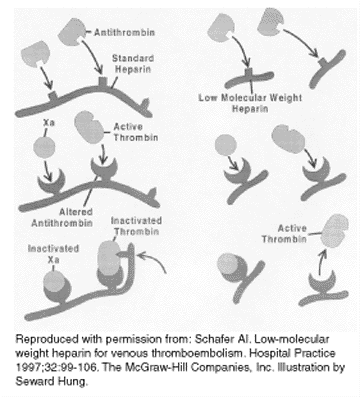
Differences also exist among the LMWHs. Comparisons are difficult because no standardization exists and their activities likely result from multiple mechanisms. The most prominent difference is that the anti-Xa to antithrombin ratio varies among the LMWHs. However, the significance of this variation is not known. Head-to-head trials comparing the different LMWHs have not been performed.
LMWHs for Unstable Angina and NSTEMI: Clinical Trials and Comparative Outcomes
Several trials have assessed the efficacy and safety of LMWHs in the management of patients with UA and NSTEMI.19,26,27 (See Table 2 by clicking here.) These trials have compared various combinations of aspirin, heparin, and LMWH. Indications for LMWHs in ACS are similar to those of heparin; both enoxaparin and dalteparin have received FDA approval for the management of ACS.
Nadroparin: The first trials comparing LMWH to standard, heparin-based regimens were small and open-labeled, and employed a randomized, single-blinded, study of 219 patients with underlying ischemic heart disease who presented with UA.19 Patients were randomized to receive intravenous heparin plus aspirin, once-daily subcutaneous nadroparin plus aspirin, or aspirin alone. The primary end point was the composite of death, AMI, or recurrent angina. Nadroparin plus aspirin reduced the number of patients with the end point from 59% in the aspirin group and 63% in the aspirin-plus-heparin group to 22% (P = 0.001).19
The FRAXIS trial compared nadroparin with heparin in 3468 patients with UA or NSTEMI.28 Patients were randomized to receive intravenous heparin for six days or twice-daily subcutaneous nadroparin for six days or for 14 days. The primary end point was the composite of cardiovascular death, AMI, or recurrent/refractory angina at 14 days. No difference in outcome was found between the treatment groups. While the 14-day regimen of nadroparin resulted in significantly more bleeding than did heparin (3.5% vs 1.6%), six days of therapy with nadroparin resulted in major bleeding in only 1.5% (see Table 2). At three months, the patients who received nadroparin for 14 days had a significantly increased incidence of cardiac events (26.2% vs 22.2% with heparin), as well as increased hemorrhage (4.0% vs 2.4% with heparin).28
Dalteparin (Fragmin): Dalteparin was the first LMWH to receive an indication for use in ACS in Europe. These trials26,30,31 used prolonged anticoagulant therapy (up to 90 days) because the risk of recurrent ischemia remains high for 6-12 weeks and because coagulant activity and thrombin generation also are increased for months after an acute event.26 This approach targeted the "rebound" or reactivation of the thrombotic process that may occur after heparin is discontinued in patients with ACS. The "rebound" may result from incomplete healing of damaged endothelial barrier at the time heparin is stopped.29 It is debatable whether the events are a true rebound or just incomplete therapy for a continuing process. It was hoped that "chronic" administration of dalteparin would confirm that low-dose, daily injections could reduce the incidence of "rebound."
FRISC Trial: The FRISC trial was a randomized comparison of dalteparin to placebo in 1506 patients who presented within 72 hours of the onset of UA or NSTEMI.26 Unlike many of the other LMWH trials, there was no comparison with heparin. The primary end point was the composite of death or AMI at six days. A secondary goal was to determine whether long-term anticoagulant therapy would provide additional benefit beyond that obtained with anticoagulation only during the acute phase.
The dalteparin group received 120 IU/kg twice daily during the "acute" phase. Patients who received dalteparin during the acute phase also received once-daily subcutaneous dalteparin (7500 IU) during the 35-45 day "chronic" phase. Dalteparin significantly reduced the frequency of death or AMI from 4.8% to 1.8% (P = 0.001) at six days; however, the difference was no longer significant at 40 days (see Table 2). The composite end point (death, myocardial infarction, revascularization, or intravenous heparin use) also was significantly decreased in the dalteparin group at 40 days. However, no significant differences between groups existed by 150 days, reflecting that long-term, once-daily dosing was inadequate for ACS.26 There was no significant difference in bleeding between the groups.
FRIC Trial: The FRIC trial was a randomized comparison of dalteparin with heparin in 1482 patients who presented within 72 hours of the onset of UA/NSTEMI.30 It is the only trial that directly compared dalteparin with heparin. The primary end point was the composite of death, AMI, or recurrent ischemia. The dalteparin group received 120 IU/kg twice daily during the six-day "acute" phase. Patients who received dalteparin during the acute phase received once-daily subcutaneous dalteparin (7500 IU) during the 35-45 day "chronic" phase. In summary, there was no significant difference in the composite outcome between the patient groups at six days (see Table 2). Both groups had a composite outcome of 12.3% at 45 days.30 Of note is that the composite end point was actually more common in those who received dalteparin. In fact, six-day mortality was significantly increased in the dalteparin group compared to the heparin group (1.5% vs 0.4%; P = 0.05). Bleeding complications were similar among the groups.
FRISC II Trial: The FRISC II trial assessed the efficacy of long-term treatment with dalteparin vs. placebo in a double-blinded, randomized comparison of 2267 patients who received either dalteparin or placebo for three months.31 (See Table 2.) As with FRISC, this trial was not a comparison with heparin. The dalteparin group received 120 IU/kg twice daily during the "acute" phase and 7500 IU twice daily (5000 IU for smaller patients) during the remaining three months. The primary end point was the composite of death or AMI at six months. There was a non-significant decrease in the primary composite end point of 6.7% and 8.0% in the dalteparin and placebo groups, respectively (P = 0.17). The difference was significant at 30 days (3.1% vs 5.9%, respectively; P = 0.002). There also was a decrease in death, AMI, or need for revascularization from 33.4% to 29.1% in the dalteparin group (P = 0.031). The differences were not sustained at six-month follow-up. Treatment with dalteparin was associated with an increased risk of major bleeding (3.3% vs 1.5%) and more hemorrhagic strokes (8 vs 0 events).31
GUSTO 4 Trial: Dalteparin was compared with heparin in a sub-study of the GUSTO 4 Trial in which 974 (13%) of the 7800 total patients received dalteparin instead of heparin.32 The focus of this trial was the comparison of a 24-hour infusion of abciximab, a 48-hour infusion of abciximab, and a placebo infusion in patients with non-ST-segment elevation ACS. Patients who received dalteparin were spread evenly among the three arms of the trial. There was no difference in the primary end point of the composite of death or AMI at 30 days among dalteparin patients in any of the three treatment arms. Major bleeding and other adverse events were comparable between the dalteparin plus abciximab group and the dalteparin alone group. Data from this major trial were recently presented at the European Society of Cardiology Congress and the full paper has not yet been published.32
Enoxaparin (Lovenox): Enoxaparin was the first LMWH to receive FDA approval for the management of ACS. This approval was based upon two positive trials in comparison with heparin (unlike the trials vs placebo as was the case for dalteparin).
The ESSENCE trial was a randomized, double-blinded study of 3171 patients with UA or NSTEMI who presented within 24 hours of symptom onset.27 (See Table 2.) Groups were treated as long as eight days with either subcutaneous enoxaparin 1 mg/kg twice-daily or intravenous heparin. The primary end point was the composite of death, AMI, or recurrent angina at 14 days.
Enoxaparin resulted in a significant reduction of the end point from 19.8% to 16.6% (P = 0.02) at 14 days and from 23.3% to 19.8% (P = 0.02) at 30 days. The most significant reduction was in the recurrence of angina. The need for urgent revascularization (coronary artery bypass or PTCA) during the 30-day study period was relatively reduced by 16% in the enoxaparin arm (from 32.2% to 27.0%; P = 0.001). The groups had comparable major bleeding events.27
This was the first major trial using a LMWH that reflected significant superiority over heparin. One-year follow-up data for 2915 of the patients (92%) found that the combined end point of death, AMI, or recurrent angina was lower in enoxaparin-treated than heparin-treated patients (32.0% vs 35.7%; P = 0.02).33
The TIMI IIB trial was a randomized comparison of 3910 patients with UA or NSTEMI.34 (See Table 2.) Patients initially were eligible if they had a significant history of CAD, EKG changes, or cardiac marker elevation. However, after 1800 patients had been enrolled, the focus was changed to include higher-risk patients by requiring that all patients have either ST-segment deviation or positive serum markers. Patients received as many as eight days of weight-adjusted intravenous heparin followed by placebo or enoxaparin (30 mg bolus, then twice-daily 1 mg/kg subcutaneous injections for up to eight days, then as many as 43 days of low-dose therapy).34 The primary end point was the composite of death, AMI, or need for urgent revascularization at eight and 43 days.
Patients receiving enoxaparin had significantly fewer end point events at eight days (12.4% vs 14.5%; P = 0.048) and at 14 days (14.2% vs 16.7%; P = 0.03). At 43 days, the beneficial effect of enoxaparin proved to be durable (end points: 17.3% vs 19.7%; P = 0.05); however, no further relative decrease in events was observed.34 Major hemorrhage was similar between groups during the acute phase; however, by day 43, it had occurred in 2.9% of those treated with enoxaparin compared with 1.5% in the placebo arm (P = 0.02).34 As was the case in ESSENCE, enoxaparin proved to be superior to heparin in TIMI IIB. However, administration of enoxaparin beyond the hospitalization phase cannot be recommended because no incremental benefit was achieved by continuing the enoxaparin treatment beyond the initial hospitalization and because of the increased risk of hemorrhage in the outpatient setting.
A meta-analysis of the ESSENCE and TIMI IIB trials found that, in comparison to heparin, enoxaparin treatment is associated with a 20% relative reduction in clinical events in patients with UA or NSTEMI. The reduction was achieved without a significant increase in the rate of major hemorrhage during the acute phase of therapy. These data support preferential use of enoxaparin over heparin and other LMWHs as the foundation antithrombin agent for patients with UA and NSTEMI.
LMWH Costs: Evidence currently supports the cost benefit of LMWHs over heparin for ACS. Cost efficacy in CAD was addressed by a pharmacoeconomic analysis of the ESSENCE trial.35 These investigators found that the use of enoxaparin vs. heparin saved $763 by hospital discharge and $1172 at 30 days. The greatest change in resource use was a decrease in coronary angioplasty. This analysis did not consider the nursing and pharmacy labor costs associated with the use of these agents, which would likely have resulted in even further savings with enoxaparin therapy.35
Management of ACS (Unstable Angina and NSTEMI) with LMWHs: Summary of Current Guidelines
Comparing the different LMWHs is difficult. Outcomes of the trials varied either because of dissimilar pharmacologic properties of the LMWHs or because the trial designs differed as to patient selection, relative doses of medication, active treatment duration, and/or the definition and assessment of end points. In addition, the trials used different durations of therapy and drug doses for both the LMWH and for heparin. Another difference among trials is that the last episode of chest pain was within 72 hours in the FRISC and FRIC trials using dalteparin, whereas chest pain was within 24 hours of onset in the enoxaparin trials, ESSENCE, and TIMI IIB. This is not likely to be of significance because more than 90% of the patients in the dalteparin trials were enrolled in less than 24 hours.
Although antithrombotic therapy is used for less than a week in patients with UA/NSTEMI, there is a rationale for more prolonged treatment since a coronary lesion thrombus lasts for several months and coagulation activity is elevated for at least three months.36 Five of the six LMWH trials (see Table 2) addressed this issue by using the agent for between eight and 90 days. Neither FRIC (dalteparin) or FRAXIS (nadroparin) found the LMWH to be superior to heparin at any primary end point, let alone any extended period. The FRISC trial (dalteparin) found no benefit to use the drug therapy beyond the acute study period.26 The FRISC II (dalteparin) trial did find superiority for the LMWH over heparin at 30 days, but it is unclear whether this occurred because of the acute phase therapy or from the chronic phase of therapy.
TIMI IIB found enoxaparin to be superior to heparin at 43 days, and there was no incremental benefit over that which existed at 14 days. Interestingly, the ESSENCE trial, which used enoxaparin only during the acute phase, found continued benefit at 30 days. However, as with TIMI IIB, there was no incremental benefit beyond that which existed at 14 days. Prolonged administration of a LMWH resulted in more major hemorrhage in three (FRAXIS, FRISC II, TIMI IIB) of the five trials that used chronic phases of administration of drug. Most of the trials had increased minor bleeding in the LMWH groups. These were mostly ecchymoses at injection sites and hematomas at vascular access sites.
Despite the difficulty in making certain comparisons among the LMWHs, some clinically relevant conclusions can be made. All of the LMWH trials involved very high-risk patients, virtually all of which had either EKG changes or NSTEMI. Nadroparin was superior to placebo in one trial19 and dalteparin was superior to placebo in both FRISC26 and FRISC II.31 These data, in addition to the finding that heparin was superior to placebo, reflect that high-risk patients should receive some heparin product, and the preferred agent is enoxaparin.
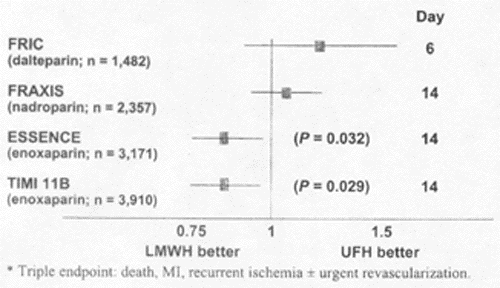
Of the four large, randomized clinical trials that compared a LMWH with heparin (FRIC, ESSENCE, TIMI IIB, and FRAXIS trials), only two trials, both using enoxaparin (ESSENCE and TIMI IIB), reflected superiority over heparin (see Figure 3). Therefore, only enoxaparin has been shown to be superior to heparin in the management of patients with UA/NSTEMI. There are not enough data to support the administration of any LMWH beyond the acute hospital phase and that chronic administration is associated with increased occurrence of major hemorrhage.
|
Figure 3. Low Molecular Weight Heparins: Effect on Triple End Points in Comparison to Heparin |
|
|
| The use of LMWH in unstable angina/NSTEMI on the triple end point of death, AMI, and recurrent ischemia with or without revascularization. Early (6 day) and intermediate outcomes of the four trials that compared LMWH and heparin: ESSENCE, TIMI IIB, FRIC, and FRAXIS. Nadroparin in FRAXIS was given for 14 days. |
| Reproduced with permission from: Braunwald E, Antman EM, Beasley JW, et al. The ACC/AHA Guidelines for Unstable Angina. J Am Coll Cardiol. 2000;36:1055. |
Many factors will affect the future use of LMWHs for ACS. Some cardiologists are concerned about using the long-acting LMWHs at the time of PCI. Another factor is the increased use of antiplatelet agents, including the glycoprotein IIb/IIIa inhibitors, thromboxane synthetase inhibitors (ridogrel), and ADP binding inhibitors (ticlopidine and clopidogrel). The interaction between these agents is unclear. Even with the rapidly expanding uses of the GP IIb/IIIa inhibitors, anticoagulation with an antithrombin agent remains mandatory in the current management of ACS. This was dramatically demonstrated in the PRISM-PLUS trial. Although the combination of low-dose tirofiban combined with heparin produced a significant reduction in death, AMI, or refractory ischemia at seven days, the tirofiban arm was discontinued by the safety monitoring committee because of an excessive mortality rate in comparison to the tirofiban-heparin or heparin-alone arms.
References
1. 1AA ACC/AHA Taskforce on practice guidelines (Committee on management of Acute Myocardial Infarction). J Am Coll Cardiol. 1996;28:1328-1428.
2. Weitz JI, Bates S. Beyond heparin and aspirin. Arch Intern Med. 2000;160:749-758.
3. Theroux P, et al. Aspirin, heparin, or both to treat acute unstable angina. N Engl J Med. 1988;319:1105-1111.
4. Lovenox. Enoxaparin Insert. Lovenox Package Insert 1999;1-1.
5. Fox I, et al. Anticoagulant activity of hirulog, a direct thrombin inhibitor, in humans. Thromb Haemost. 1993; 69:157-163.
6. Ambrose JA, Dangas G. Unstable angina: Current concepts of pathogenesis and treatment. Arch Intern Med. 2000;160:25-37.
7. Hull RD, Pineo GF. Hirudin versus heparin and low-molecular-weight heparin: And the winner is . . . J Lab Clin Med. 1998;132:171-174.
8. Cannon C, Braunwald E. Hirudin: Initial results in acute myocardial infarction, unstable angina and angioplasty. J Am Coll Cardiol. 1995;25:30S-37S.
9. GUSTO IIb Investigators. A comparison of recombinant hirudin with heparin for the treatment of acute coronary syndromes. N Engl J Med. 1996;335:775-782.
10. OASIS-2 Investigators. Effects of recombinant hirudin (lepirudin) compared with heparin on death, myocardial infarction, refractory angina, and revascularization procedures in patient with acute myocardial ischemia without ST elevation: A randomized trial. Lancet. 1999;353:423-424.
11. Bittl JA, et al. Treatment with bivalirudin (HIRULOG) as compared with heparin during coronary angioplasty for unstable or postinfarction angina. N Engl J Med. 1995;333:764-769.
12. Serruys PW, et al. A comparison of hirudin with heparin in the prevention of restenosis after coronary angioplasty. N Engl J Med. 1995;333:757-763.
13. The RISC Group. Risk of myocardial infarction and death during treatment with low-dose aspirin and intravenous heparin in men with unstable coronary artery disease. Lancet. 1990;336:827-830.
14. Theroux P, et al. Aspirin versus heparin to prevent myocardial infarction during the acute phase of unstable angina. Circulation. 1993;88:2045-2048.
15. Theroux P, et al. Reactivation of unstable angina after the discontinuation of heparin. N Engl J Med. 1992; 327:141-145.
16. Cohen M, et al. Usefulness of antithrombotic therapy in resting angina pectoris or non-Q-wave myocardial infarction in preventing death and myocardial infarction (a pilot study from the Antithrombotic Therapy in Acute Coronary Syndromes Study Group). Am J Cardiol. 1990;66:1287-1292.
17. Cohen M, et al. Combination antithrombotic therapy in unstable rest angina and non-Q-wave infarction in non-prior aspirin users: Primary end points analysis from the ATACS Trial. Circulation. 1994;89:81-88.
18. Holdright D, et al. Comparison of the effect of heparin and aspirin versus aspirin alone on transient myocardial ischemia and in-hospital prognosis in patients with unstable angina. J Am Coll Cardiol. 1994;24: 39-45.
19. Gurfinkel EP, et al. Low molecular weight heparin versus regular heparin or aspirin in the treatment of unstable angina and silent ischemia. J Am Coll Cardiol. 1995;26:313-318.
20. Oler A, et al. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. JAMA. 1996;276: 811-815.
21. Fareed J, et al. Low-molecular-weight heparins: Pharmacologic profile and product differentiation. Am J Cardiol. 1998;82:3L-10L.
22. Makris M, et al. Poor reversal of low molecular weight heparin by protamine. Br J Haematol. 2000;108: 884-885.
23. Hickey A. Amy D. Hickey, RN, Clinical Nurse Specialist. Personal Communication, 1998.
24. Warkentin TE, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332:1330-1335.
25. Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med. 1996;101: 502-507.
26. FRISC study group. Low-molecular-weight heparin during instability in coronary artery disease. Lancet. 1996;347:561-568.
27. Cohen M, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. N Engl J Med. 1997;337:447-452.
28. The FRAXIS Study Group. Comparison of two treatment durations (6 days and 14 days) of a low molecular weight heparin with a 6 day treatment of unfractionated heparin in the initial management of unstable angina or non-Q-wave myocardial infarction: FRAX.I.S. (FRAxiparine in Ischaemic Syndrome). Eur Heart J. 1999;20:1553-1562.
29. Granger CB, et al. Rebound increase in thrombin generation and activity after cessation of intravenous heparin in patients with acute coronary syndromes. Circulation. 1995;91:1929-1935.
30. Klein W, et al. Comparison of low-molecular-weight heparin with unfractionated heparin acutely and with placebo for 6 weeks in the management of unstable coronary artery disease. Circulation. 1997;96:61-68.
31. FRISC II Investigators. Long-term low-molecular mass heparin in unstable coronary-artery disease: FRISC II prospective randomized multicentre study. Lancet. 1999;354:701-707.
32. Simoons M, Wallentin L. GUSTO 4 acute coronary syndromes. Presentation: European Society of Cardiology Congress, 2000.
33. Goodman S, et al. Antithrombotic therapy in acute coronary syndromes. Eur Heart J. 1998;199 (Suppl l):477.
34. Antman EM, et al. Enoxaparin prevents death and cardiac ischemic events in unstable angina/non-Q-wave myocardial infarction. Circulation. 1999;100: 1593-1601.
35. Mark DB, et al. Economic assessment of low-molecular-weight heparin (enoxaparin) versus unfractionated heparin in acute coronary syndrome patients results from the ESSENCE randomized trial. Circulation. 1998;97:1702-1707.
36. Wallentin L. Efficacy of low-molecular-weight heparin in acute coronary syndromes. Am Heart J. 2000;139(2 Part 2):S29-S32.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.