The Evidence-based Approach to Neurologic Emergencies: Part II: Hemorrhagic Stroke and Traumatic Brain Injury
The Evidence-based Approach to Neurologic Emergencies: Part II: Hemorrhagic Stroke and Traumatic Brain Injury
Author:
Ademola Adewale, MD, FAAEM, Assistant Professor of Emergency Medicine, Florida State University College of Medicine, Director of Research and Medical Simulation, Florida Hospital Emergency Medicine Residency Program, Orlando, FL.
Peer Reviewer:
Bentley J. Bobrow, MD, Associate Professor of Emergency Medicine, Emergency Medicine Department, Maricopa Medical Center, Phoenix, AZ.
The first part of this series dealt with guidelines for the care of ischemic stroke. This issue applies the same principles of evidence-based medicine to hemorrhagic stroke and traumatic brain injury. Hemorrhagic stroke is less common than ischemic stroke, but can be more devastating. Each of us has witnessed a patient who arrives at the emergency department (ED) with relatively mild complaints and quickly deteriorates or has a seizure. Such patients often require immediate resuscitative care, including intubation before the true diagnosis, and prognosis, is clear. Like ischemic stroke, there remains controversy about the use of aggressive neurosurgical evacuation of the clot versus simply allowing the bleeding to be reabsorbed by the brain. While there are occasional patients who recover, albeit with some deficits, many individuals with significant hemorrhagic stroke eventually die or no longer can lead an independent life.
Like hemorrhagic stroke, traumatic brain injury can lead to devastating consequences. While there is a lot of attention at the moment to mild traumatic brain injury, this issue deals with severe injuries. While we often feel somewhat helpless when we see the patient with a devastating brain injury, current guidelines may provide some guidance to reduce swelling and prevent further ischemia to vulnerable brain tissue.
In addition to providing an update on the latest care for patients with these serious injuries, this issue serves as a reminder that the brain is a vulnerable organ. Trapped within the rigid skull, there is little room for the brain to swell. Brain tissue loss, particularly in the adult, is permanent. Our goal is to reduce swelling and preserve as much function as possible.
Sandra M. Schneider, MD, FACEP, Editor
Hemorrhagic Stroke
Hemorrhagic stroke, or intracerebral or intraparenchymal hemorrhage, accounts for 10-15% of all strokes, and it is associated with significant mortality when compared to ischemic stroke.1 This subset of stroke is divided into two categories: intracerebral (ICH) and subarachnoid hemorrhage (SAH). The rupture of deep, penetrating arteries with resultant bleeding into the brain tissue is referred to as ICH, which accounts for about 10% of all strokes. The most common cause of ICH is hypertension, and a significant proportion of patients with the diagnosis of ICH have a history of hypertension. Collectively, aneurysm, hypertension, and vascular malformation account for almost 80% of ICH. Annually, about 70,000 Americans suffer from this disabling disease.2
The other subset, SAH, affects about 30,000 people annually and it is mostly due to the rupture of an aneurysm or arteriovenous malformation into the subarachnoid space.2 The most commonly seen aneurysms in intracranial bleeding are Berry and saccular aneurysms. Hemodynamic stress and acquired or congenital vessel wall weakness are the common causes of Berry aneurysm. On the other hand, saccular aneurysms tend to form at the bifurcation of vessels, and almost 90% occur at the anterior circulation. Most, but not all, patients with SAH present with the sudden onset of the worst headache of their life, which may be accompanied by nausea, vomiting, and neck stiffness. Nonenhanced CT of the head often shows the presence of a subarachnoid hemorrhage (see Figure 1), especially if done early after the onset of headache. Lumbar puncture may be necessary to make the diagnosis, especially if the headache onset was more than 6 hours previously.
Figure 1: CT of Subarachnoid Hemorrhage
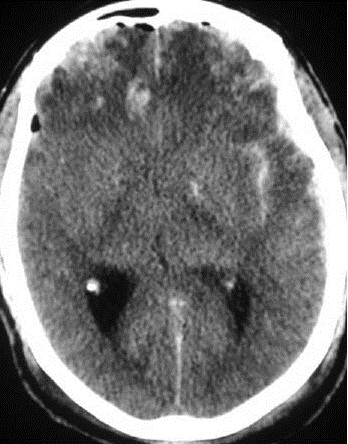
Nonenhanced CT of a patient with a subarachnoid hemorrhage
Reprinted from Menaker J, Scalea TM. Traumatic brain injury. Trauma Reports 2009;10:4.
Regardless of the subset, these patients present with a myriad of symptoms, such as focal neurologic deficit, headache, nausea, vomiting, dizziness, and altered level of consciousness. The evaluation and management of patients with hemorrhagic stroke will be discussed in the context of the case below.
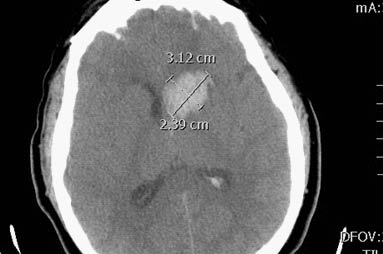
A 48-year-old male arrived at the ED at 2:30 p.m. with a severe headache, nausea, and vomiting. While being checked-in at the ambulance triage, the patient began vomiting profusely, had an acute change in mental status, and a grand mal seizure. The patient was taken to the resuscitation area where intravenous access was established, 1 mg of intravenous lorazepam was given, and the seizure terminated. The patient was subsequently intubated for airway protection. The initial vital signs when placed on a monitor were: blood pressure 240/120, pulse 112, and oxygen saturation of 100% on a ventilator. The patient was loaded with 1 gram of fosphenytoin and sent for an emergent CT scan of the brain. The CT scan confirmed an intracerebral hemorrhage. (See Figure 2.)
Figure 2: Nonenhanced CT of the Patient Showing Intracerebral Hemorrhage in the ACOM and MCA Distribution
Therapeutic Intervention
The likelihood of early worsening of neurologic status and cardiopulmonary instability is very high in this patient population. Determining the neurologic status using a grading scale such as the Glasgow Coma Scale (GCS) is important. A study by Broderick et al3 demonstrated that the volume of the ICH and the GCS on admission are the most powerful predictors of death in 30 days.
The ED medical treatment of ICH patients focuses on preventing the progression of the bleeding by the following: acute blood pressure treatment, treatment of increased intracranial pressure (ICP), brain supportive therapy (ICP and CPP), glucose control, prevention and treatment of seizure, temperature control, and monitoring of neurologic and cardiopulmonary functions.
Blood Pressure Management. Management of blood pressure in ICH requires understanding cerebral autoregulation. An uninjured brain autoregulates its blood flow by altering cerebral vascular resistance to provide constant flow regardless of blood pressure by maintaining adequate cerebral perfusion pressure (CPP). The CPP is the difference between the mean arterial pressure (MAP) and the ICP. In the event of ICH, the cerebral autoregulatory capacity is compromised, and the brain becomes susceptible to fluctuation in blood pressure. Significantly elevated blood pressure may lead to increased hematoma volume.
The American Heart Association (AHA) and the American Stroke Association (ASA) 2007 guidelines for acute blood pressure management in ICH recommended a target systolic blood pressure of 180 mm Hg. However, preliminary data from two recent studies (INTERACT4 and ATACH5) prompted the AHA/ASA to modify their recommendation in the 2010 guidelines. The current guidelines suggest that acute lowering of SBP to 140 mm Hg is probably safe in patients with ICH with a presenting SBP between 150-220 mm Hg6 (this is a class IIa recommendation). The treatment guideline is stated below2,6:
1. If SBP is > 200 mm Hg or MAP is > 150 mm Hg, consider aggressive reduction with continuous infusion of IV antihypertensive.
2. If SBP is > 180 mm Hg or MAP is > 130 mm Hg, and the patient may have elevated ICP, consider monitoring ICP and reducing BP with intermittent or continuous IV medication while maintaining CPP ≥ 60 mm Hg.
3. If SBP is > 180 mm Hg or MAP is > 130 mm Hg, and there is no evidence of elevated ICP, consider using intermittent or continuous IV antihypertensive to achieve a modest reduction in BP (target BP of 160/90 mm Hg or MAP of 110 mm Hg).
All of the above guidelines are class IIb, level of evidence C recommendations.
Returning to the patient, on identification of ICH on the CT scan, the patient was started on intravenous nicardipine drip at 5 mg/hour and titrated to a goal of SBP < 160 mm Hg. In the interim, the patient had received anti-seizure medication, and was sedated on a benzodiazepine drip.
Seizure Control. Seizures occur commonly in patients with ICH. Lack of immediate seizure control may contribute to ICP elevation. A study by Passero et al7 evaluated 761 patients and noted that early seizure occurred in about 4.2% of the patients, and 8.1% had seizures within 30 days. Vespa et al8 evaluated a cohort of patients with ICH undergoing continuous EEG monitoring and noted that seizure occurred in about 28% of the patients during the initial 72 hours of admission. They concluded that seizures related to ICH are often nonconvulsive and tend to be associated with higher NIHSS scores, the presence of midline shift, and portend a bad outcome.
According to the AHA/ASA (class I, level of evidence B), clinical seizures should be controlled promptly with intravenous benzodiazepine, followed by intravenous fosphenytoin or phenytoin.
Management of Elevated ICP. The hematoma from ICH displaces brain tissue secondary to hematoma growth and perihematomal edema. This process will potentially lead to elevated ICP. The measurement of ICP is beyond the purview of emergency medicine; however, steps could be taken in the emergency department to prevent the worsening of ICP.
Several modalities that are presently used to combat elevated ICP include: hyperventilation, head of bed elevation, neuromuscular blockade, osmotic therapy, systemic cooling, and induced coma state.
Hyperventilation. This is the most commonly utilized and effective way to rapidly reduce ICP. However, it reduces ICP by causing cerebral vasoconstriction with concomitant reduction in cerebral blood volume and flow. A study by Fortune et al9 demonstrated that decreasing the arterial PCO2 to 26 mm Hg in eight healthy individuals decreased cerebral blood volume by 7.2%. Hayes et al10 also demonstrated that the correlation between PaCO2 and ICP is not linear and that a significant benefit is noted between PaCO2 values of 30 and 50 mm Hg in humans. A recent study by Carrera et al11 that evaluated the effect of spontaneous hyperventilation on brain tissue hypoxia (BTH) demonstrated that even at a PCO2 level of 35-44 mm Hg, brain tissue hypoxia increased by 15.8%. Furthermore, when PCO2 was reduced to less than 25 mm Hg, the level of BTH increased to almost 34%. The study concluded that the risk of BTH in critical brain-injured patients increases when ETCO2 values are reduced. Although hyperventilation is effective in lowering ICP, it does have some drawbacks: the transient nature of its effect, concomitant decrease in cerebral blood flow, brain tissue hypoxia, and rapid normalization of PCO2 that leads to rebound increase in ICP.
According to Stocchetti et al,12 controversies exist in the use of hyperventilation. The careful use of hyperventilation for short-term control of ICP is a useful therapeutic tool and the target levels for CO2 in hyperventilation as a modality to reduce ICP should be between 30 to 35 mm Hg. In addition, the American Stroke Association agrees with the range of 30 to 35 mm Hg, and recommended against any level below that target number.
Head of Bed Elevation. Elevation of the head of bed to 30 degrees has been shown to improve jugular venous outflow and lower ICP.2 The head should be midline, and head turning to either side should be avoided. In patients who are hypovolemic, elevation of the head of the bed may be associated with a fall in blood pressure and an overall fall in CPP; therefore, care must be taken initially to exclude hypovolemia. A study by Rosner et al13 demonstrated that for every 10-degree elevation in the head of the bed, the average ICP decreases by 1 mm Hg with a simultaneous decrease in CPP by 2 to 3 mm Hg. The paper further demonstrated that zero degree head elevation maximizes the CPP. They recommended that in order to avoid compromise of CPP, if the head of the bed has to be elevated to reduce ICP, adequate hydration should be maintained and blood pressure lowering should be used judiciously.
Schwarz et al14 looked at the effect of body position on intracranial pressure and cerebral perfusion in patients with large hemispheric stroke. Their data demonstrated that blood pressure, CPP, and ICP fell as the head of the body was raised from 0 to 30 degrees. Despite the fact that ICP fell on average from 13 mm Hg to 11 mm Hg at 30 degrees, the mean BP dropped from a baseline of 90 mm Hg to 76.1 mm Hg at 30 degrees. The paper concluded that the supine position was preferable with regard to CPP, optimal body positioning should be individualized, and that routine use of 30-degree elevation could not be endorsed.
In summation, to accomplish the standard recommendation of "head of bed elevation of 30 degrees" that has been shown to reduce ICP, blood pressure lowering should be targeted to the MAP that will allow the CPP range of ≥ 70 mm Hg.
Osmotic Therapy. Mannitol has been used for ICP reduction for many years. It is an osmotic diuretic that has a significant effect on ICP, brain metabolism, and cerebral blood flow. It decreases blood viscosity, expands circulating volume, and, thus, increases cerebral blood flow, CPP, and oxygen transport to the brain, while decreasing ICP through cerebral autoregulation. It is best utilized as a bolus when acute reduction in ICP is needed.
In the case example, there were suspicions of elevated ICP. The patient was hyperventilated to a PCO2 of 30 to 35 mm Hg using the capnometer to monitor the PCO2 level, the head of bed was elevated, blood pressure maintained at SBP ≤ 160 mm Hg, and DBP of ≤ 90 mm Hg (MAP of 110-115 mm Hg), and the neurosurgeon was consulted. The patient was subsequently taken to the operating room for emergent clot evacuation.
Traumatic Brain Injury
The CDC Injury Prevention and Control on TBI estimates that each year about 1.7 million people sustain traumatic brain injury (TBI). Of these, 52,000 will die, 275,000 will be hospitalized, and almost 1.365 million, or nearly 80%, are treated and released from emergency departments.15 Presently, TBI is the third contributing factor (30.5%) of all injury-related deaths in the United States, and adults aged 75 years and older have the highest rates of TBI-related hospitalization and deaths.15
The definition of TBI varies. In order to streamline the definition, the Demographics and Clinical Assessment Working Group of the International and Interagency Initiative Toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health proposed the following definition: "TBI is defined as an alteration in brain function, or other evidence of brain pathology, caused by an external force."
Traumatic brain injury includes concussion and mild TBI (jolt or blow to the head resulting in transient change in mental status or loss of consciousness), and severe TBI (significant blow to the head or penetrating injury that results in severe alteration in the level of consciousness for an extended period and amnesia after injury). Approximately 75% of TBIs that occur each year are concussions or other forms of TBI.16 For the purpose of this paper, the emphasis will be on the severe form of TBI.
Severe TBI has a significant impact on the patient, his or her family, the economy, and the society as a whole. It is estimated that in 2010, the total direct and indirect medical financial burden of TBI was $76.5 billion. In addition, severe TBIs that require hospitalizations or result in fatalities account for approximately 90% of these costs.17,18 There are two subtypes of severe TBI:
closed injury, caused by movement of the brain within the skull as a result of a motor vehicle collision, a fall, or a blow to the head;
penetrating injury, caused by penetrating injury to the skull such as impalement or firearms-related injuries.
According to the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health database, almost half of all TBIs are due to automobile, motorcycle, bicycle, and pedestrian accidents. These accidents are the major cause in patients younger than age 75 years. For patient older than 75 years, falls are the most common cause of TBI. Furthermore, about 20% of TBI occurs as a result of violence, assaults with firearms, and child abuse. About half of these cases involve alcohol.19
The evaluation and management of patients with TBI starts with the prehospital personnel. A study conducted by Hartl et al evaluated the impact of direct transport of TBI patients within an organized state trauma system on mortality reduction. The study concluded that there is class II evidence that demonstrates a 50% increase in mortality with a transfer to non-trauma centers. They recommended that patients with severe TBI should be transported to a level I or II trauma center with capabilities defined by the guidelines for prehospital management of TBI.20
In the prehospital setting, apply the PHTLS algorithm. To ensure oxygenation, assess the airway, prevent hypoxemia keeping the oxygen saturation > 90%, and ensure adequate ventilation by using the end tidal CO2 monitor when available. Hyperventilation should be avoided, particularly in the prehospital setting. Maintain adequate circulation to ensure adequate blood pressure (strictly avoiding and treating SBP < 90 mm Hg) to maintain adequate CPP. Maintaining the SBP greater than 90 mm Hg and the MAP ≥ 110 mm Hg improves survival. Several studies21-23 have shown that hypotension occurs in about 16-20% of TBI patients and this hypotension is an independent predictor of worse outcome. In fact, a single episode of SBP less < 90 mm Hg is associated with a doubling of mortality in TBI.24
The Glasgow Coma Scale (GCS) has been used as the objective measure of level of consciousness since Teasdale and Jennet developed it in 1974.25 The GCS has since been shown to have a high inter- and intra-rater reliability by several studies,26,27 and it has been shown to correlate well with outcome. A prospective study performed by Narayan28 demonstrated a positive predictive value of 77% for poor outcome with a GCS score of 3-5, and 26% for a GCS 6-8. In another study involving more than 700 patients with closed head injury from the Trauma Coma Database (TCDB) to determine the correlation between the initial GCS score and outcome, the mortality rate for the initial post-traumatic GCS of 3 was 78.4%, initial GCS 4 was 55.9%, and GCS of 5 was 40.2%.29 Based on the available evidence, the initial GCS screening should be performed promptly in the field or in the ED before sedation or analgesia for outcome prediction purposes.
After screening, the management of these patients should follow the Brain Trauma Foundation (BTF) in-hospital severe TBI guidelines. A study conducted by the CDC demonstrated that adopting the guidelines reduces mortality by 50%, saves approximately $288 million in medical and rehabilitation costs, and saves about $3.8 billion in the societal lifelong cost of the care of TBI patients.30 For the purpose of this paper, discussion of these guidelines will be limited to those involving the emergency department (blood pressure and oxygenation, hyperosmolar therapy, prophylactic hypothermia, indication for ICP monitoring, hyperventilation, seizure prophylaxis, and steroid utilization).
Hyperventilation. Hyperventilation has been the cornerstone for rapid ICP reduction in TBI for decades. Available evidence shows that the cerebral vasoconstriction and reduction in cerebral blood volume (CBV) caused by reduced PaCO2 from hyperventilation might lead to a decrease in cerebral blood flow (CBF) to an ischemic level and thereby compromise patient outcome. There is increasing evidence to support this deleterious effect. A recent study by Cole et al31 demonstrated that the acute CBF reduction from hyperventilation leads to increased compromise in cerebral oxygen metabolism, exhausts physiologic reserves, and compromises cerebral oxidative metabolism.
Another review by Curley et al32 reexamined the rationale for the use of hypocapnia in acute brain injury and evaluated the evidence for therapeutic and deleterious effects. The review demonstrated that the effect of sustained hypocapnia on CBF decreases progressively as a result of buffering mechanism, and subsequent normocapnia can lead to rebound cerebral hyperemia with a potential increase in ICP. The review concluded that hypocapnia can cause harm and should be strictly limited to emergent management of life-threatening ICP pending definitive measures.
The BTF surmises that independent of hyperventilation, CBF can drop to a dangerous level in TBI patients, and the introduction of hyperventilation could further decrease CBF and contribute to cerebral ischemia. The BTF currently has no level I or II recommendation on the use of hyperventilation to reduce ICP. The foundation, however, has these three level III recommendations:
hyperventilation is recommended as a temporizing measure for reducing ICP;
hyperventilation should be avoided in the first 24 hours after injury when CBF is often critically reduced;
when hyperventilation is used, the jugular venous oxygen saturation (SjO2) or brain tissue oxygen tension (PbrO2) measurements are recommended to monitor oxygen delivery.
Blood Pressure. Hypotension at any time during the prehospital period or in-hospital is a predictor of poor outcome. A single prehospital hypotension of SBP < 90 mm Hg is one of the most powerful predictors of poor outcome. A study by Manley et al24 concluded that, "hypotension during the initial phase of resuscitation is significantly associated with increased mortality following brain injury, even if for a brief period." According to the BTF guidelines recommendation level II, "blood pressure should be monitored and hypotension (SBP < 90 mm Hg) should be avoided."
Oxygenation. Hypoxemia to the injured brain might lead to secondary brain insult and may also be an indicator of low CPP below the normal threshold. Prospective analysis of data collected from the Trauma Coma Database (TCDB) revealed that the incidence of hypoxemia was 22.4% in severe TBI patients and it was significantly linked to an increased morbidity and mortality.23,33 Clinical trials involving the effects of hypoxemia on TBI are difficult to conduct due to ethical concerns. Hence, there is no evidence to make any recommendations on the threshold of hypoxemia to avoid.
The BTF has no level I or II recommendations on hypoxemia. However, a level III recommendation stipulates that oxygenation should be monitored and hypoxia (PaO2 < 60 mm Hg or O2 saturation < 90%) should be avoided.
Hyperosmolar Therapy. Presently, mannitol and hypertonic saline are the commonly utilized hyperosmolar agents in severe TBI. Mannitol has been used for the control of elevated ICP and in reversing cerebral edema for decades; however, its effectiveness in prolonged utilization in the management of severe TBI is unknown, and many controversies regarding mannitol use exist.
A review of the Cochrane Database summaries by Wakai et al34 suggested that prolonged use of mannitol may actually induce elevated ICP. A study conducted by Lin K et al35 evaluating the early response of mannitol infusion in traumatic brain injury demonstrated that dynamic effect can occur within 5-10 minutes after mannitol infusion, and its greatest effect was seen in TBI patients with ICP ≥ 20 mm Hg. Another study conducted by Sorani et al,36 characterizing the dose–response relationship between mannitol and ICP in TBI concluded that the effect of mannitol on ICP is dose-dependent, and higher doses provide a more durable reduction in ICP.
Based on the available body of evidence on mannitol, the BTF concluded that mannitol is effective for control of raised ICP at doses of 0.25 mg/kg to 1 g/kg body weight (level II recommendation). They cautioned, however, that arterial hypotension should be avoided. Furthermore, the foundation recommended restricting mannitol use prior to ICP monitoring to a subset of patients with signs of transtentorial herniation or progressive neurologic deterioration not attributable to extracranial causes (level III).30
Hypertonic saline (HTS) is another common hyperosmolar agent used in acute lowering of elevated ICP. Munar et al37 evaluated cerebral hemodynamic changes with the infusion of 7.2% HTS in patients with TBI and elevated ICP. They demonstrated that there was a decrease in ICP that was associated with increased osmolality caused by the HTS. In 2009, the National Heart, Lung and Blood Institute (NHLBI) of the National Institutes of Health terminated enrollment into the largest clinical trial evaluating the effect of HTS in severe TBI. After the review of data from more than 1000 participants, the NHLBI determined that the HTS solution was not superior to standard treatment with normal saline. The BTF summary published in 2007 suggested that current evidence at the time was not strong enough to make a recommendation on the use of HTS, concentration, and method of administration in treatment of increased ICP.
A recently published meta-analysis in March 2011 comparing HTS to mannitol for the treatment of elevated ICP concluded that HTS is more effective than mannitol for the treatment of elevated ICP. The authors argued that HTS may be superior to the current standard of care and recommended a large, multicenter, randomized trial to definitely establish the first-line therapy for ICP lowering.38
Presently, controversies exist regarding the use of HTS in TBI. Its utility is based on institutional experience and practice patterns.
Prophylactic Hypothermia. During the past decade, mounting evidence to suggest the use of prophylactic hypothermia has accumulated. Studies have shown that a relatively small variation in temperature may alter cerebral hemodynamics, calcium-dependent intracellular signaling, excitotoxicity, inflammatory cerebral edema, and apoptosis.39-41
Understanding these physiologic events explains why cooling of the brain may slow down the progression of secondary injuries. However, the BTF does not have a level I or II recommendation for the utilization of prophylactic hypothermia in severe TBI. The foundation's level III statement indicates that preliminary findings demonstrate that a greater decrease in mortality risk was observed when target temperature was maintained for more than 48 hours.30 These recommendations, however, were provided in 2007.
Several more current exhaustive meta-analyses since then have produced contradictory findings. A review by Dietrich et al in 201042 identified the need to clearly define the therapeutic window and the duration of hypothermic treatment for specific patient populations. They concluded that currently "hypothermic TBI treatment remains experimental and additional experimental and clinical studies must continue to evaluate how best to use this treatment strategy in severe TBI patients."42
In contrast, a quantitative systematic review published in July 2010 by Fox et al43 evaluated two cooling categories: short term (≤ 48 hours), and long term or goal directed (> 48 hours and/ or continuous until normalization of ICP). The study demonstrated that the best available evidence supports the early utilization of prophylactic mild to moderate hypothermia in patients with severe TBI (GCS ≤ 8) to decrease mortality and improve rates of good neurologic recovery. The authors further recommended that treatment should be commenced promptly (in the emergency department after CT scan of the brain), regardless of initial ICP or before it is measured.34
Therapeutic hypothermia definitely has potential in the treatment of TBI to minimize the effects of secondary brain insults. However, more clinical studies are needed to clearly delineate the appropriate protocol for its utilization. One such study is the Prophylactic Hypothermia Trial to Lessen Traumatic Brain Injury (POLAR-RCT) in Australia and New Zealand. This is a phase III study investigating the effects on outcomes of early cooling of patients with severe TBI. The study is currently ongoing, and the findings may shed more light on the use of prophylactic hypothermia in TBI.
Post-traumatic epilepsy or seizures (PTE) are potential neurologic complications of TBI and they are reported in almost 50% of survivors.44 Post-traumatic epilepsy could be immediate (within 24 hours of injury), early (within one week), and late (more than a week after injury). In TBI patients, PTE is one of the important factors that prevents this population from returning to their pre-existing employment and lifestyle.45 The risk factors identified for developing PTE are: Glasgow Coma Scale < 10, cortical contusion, depressed skull fracture, intracerebral, subdural, and epidural hematoma, penetrating head injury, and seizure within 24 hours of injury.46-48
Table 1: Risk Factors for Stroke
|
Modifiable |
Not Modifiable |
|
|
Presently, no pharmacological or non-pharmacological therapies exist that could be considered as anti-epileptogenic for PTE. A study by Temkin49 reported that clinical trials of conventional anti-epileptic drugs have not shown any effects in protecting against development of PTE. However, several studies50,51 have demonstrated the beneficial effect of phenytoin in protecting against the development of early PTE. These same studies also show that phenytoin has no effect in preventing late PTE. In contrast, several other studies50,51 evaluating phenobarbital, phenytoin, and valproic acid found no effects in preventing PTE.
The Brain Trauma Foundation concluded that there are insufficient data to support a level 1 recommendation for anti-seizure prophylaxis for post-traumatic epilepsy. Furthermore, the prophylactic use of phenytoin and valproate is not recommended for preventing late post-traumatic seizures (level II). However, owing to some studies that show the benefit of anti-epileptic drugs in decreasing the incidence of early PTE, the BTF supports the use of anti-epileptics for early PTE (within 7 days of injury).
Figure 3: Cerebral Contusion
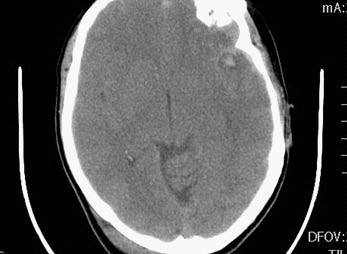
This image shows a 43-year-old male who was assaulted, showing cerebral contusion with bleed diagnosed with traumatic brain injury.
Corticosteroids. Steroids have been widely utilized in the treatment of severe traumatic brain injury based on the assumptions that they reduce intracranial pressure. However, the majority of the body of evidence suggests a deleterious effect of steroids. A multicenter study performed by Roberts et al52 evaluating intravenous methylprednisolone in more than 10,000 patients with GCS < 14 within 8 hours of injury was halted after 62 months when the interim analysis by the data monitoring committee demonstrated a clear deleterious effect. Another study by Watson et al53 in a prospective cohort of more than 400 patients demonstrated a 74% increase in the risk of first late seizures.
A Cochrane Database Systematic Review by Alderson et al54 titled "Corticosteroids for Acute Traumatic Brain Injury" demonstrated a significant increase in the number of deaths in patients given corticosteroids. Based on this finding, the Cochrane Database suggests that steroids should no longer be routinely used in people with traumatic brain injury. In addition to the above evidence, the BTF has a level I recommendation against the use of steroids in TBI. The recommendation states that "the use of steroids is not recommended for improving outcome or reducing ICP. In patients with moderate or severe TBI, high-dose methylprednisolone is associated with increased mortality and it is contraindicated."
Special Consideration
Traumatic Brain Injury or Hemorrhage While on Dabigatran (Pradaxa). Dabigatran is a direct thrombin inhibitor for the prevention of stroke or thrombosis in patients with atrial fibrillation. The half-life of the drug is 12-14 hours in patients with normal renal function. In patients with renal impairment, the drug is dialyzable with almost 60% removed after 3 hours. The concern with this drug is the lack of antidote for potentially induced coagulopathy. Fresh frozen plasma or vitamin K has no effect on reversing the effect of the drug. Prothrombin complex concentrate has been discussed as a potential antidote for dabigatran reversal. However, a recent study by Eerenberg ES et al55 demonstrated that prothrombin complex concentrate has no effect in reversal of dabigatran coagulopathy.
References
1. Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics 2009 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009;119:480-486.
2. Broderick J, Connolly S, Feldmann E, et al. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: A guideline from the American Heart Association/ American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007;38:2001-2023.
3. Broderick JP, Brott TG, Duldner JE, et al. Volume of intracerebral hemorrhage: a powerful and easy-to-use predictor of 30-day mortality. Stroke 1993;24:987–993.
4. Anderson CS, Huang Y, Arima H, et al; INTERACT Investigators. Effects of early intensive blood pressure-lowering treatment on the growth of hematoma and perihematomal edema in acute intracerebral hemorrhage: The Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trial (INTERACT). Stroke 2010;41:307-312.
5. Antihypertensive Treatment of Acute Cerebral Hemorrhage (ATACH) Investigators. Antihypertensive treatment of acute cerebral hemorrhage. Crit Care Med 2010;38:637-648.
6. Morgenstern LB, Hemphill JC III, Anderson C, et al; American Heart Association Stroke Council and Council on Cardiovascular Nursing. Guidelines for the management of spontane-ous intracerebral hemorrhage: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2010;41: 2108-2129.
7. Passero S, Rocchi R, Rossi S, et al. Seizures after spontaneous supratentorial intracerebral hemorrhage. Epilepsia 2002;43:1175-1180.
8. Vespa PM, O'Phelan K, Shah M, et al. Acute seizures after intracerebral hemorrhage: A factor in progressive midline shift and outcome. Neurology 2003;60:1441–1446.
9. Fortune JB, Feustel PJ, Graca L, et al. Effect of hyperventilation, mannitol, and ventriculostomy drainage on cerebral blood flow after head injury. J Trauma 1995;39:1091-1097.
10. Hayes TM, Tindall GT. Effects of altering arterial carbon dioxide pressure on internal carotid blood flow and cerebrospinal fluid pressure in man. Surg Forum 1969;20:421-424.
11. Carrera E, Schmidt JM, Fernandez L, et al. Spontaneous hyperventilation and brain tissue hypoxia in patients with severe brain injury. J Neurol Neurosurg Psychiatry 2010;81:793-797.
12. Stocchetti N, Maas AI, Chieregato A, van der Plas AA. Hyperventilation in head injury: A review. Chest 2005;127:1812.
13. Rosner MJ, Coley IB. Cerebral perfusion pressure, intracranial pressure, and head of bed elevation. J Neurosurg 1986;65:636-641.
14. Schwarz S, et al. Effects of body position on intracranial pressure and cerebral perfusion in patients with large hemispheric stroke. Stroke 2002;33:497-501.
15. Faul M, Xu L, Wald MM, et al. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010.
16. Centers for Disease Control and Prevention (CDC), National Center for Injury Prevention and Control. Report to Congress on mild traumatic brain injury in the United States: Steps to prevent a serious public health problem. Atlanta (GA): Centers for Disease Control and Prevention; 2003.
17. Finkelstein E, Corso P, Miller T, et al. The Incidence and Economic Burden of Injuries in the United States. New York (NY): Oxford University Press; 2006.
18. Coronado, McGuire, Faul, Sugerman, Pearson. The Epidemiology and Prevention of TBI (in press) 2012.
19. National Institute of Neurological Disorders and Stroke/National Institute of Health. Bethesda, Maryland. NIH Publication No. 02-158, September 2002.
20. Hartl R, Gerbeer LM, Iacono L, et al. Direct transport within an organized state trauma system reduces mortality in patients with severe traumatic brain injury. J Trauma 2006;60:1250-1256.
21. Garner A, Crooks J, Lee A, et al. Efficacy of prehospital critical care teams for severe blunt head injury in the Australian setting. Injury, Int J Care Injured 2001;32:455-460.
22. Hill DA, Abraham KJ, West RH. Factors affecting outcome in the resuscitation of severely injured patients. Aust N Z J Surg 1993;63:604-609.
23. Marmarou A, Anderson RL, Ward JD, et al. Impact of ICP instability and Hypotension on outcome in patients with severe head trauma. J Neurosurg 1991;75:S159-S166.
24. Manley G, Knudson M, Morabito D, et al. Hypotension, hypoxia, and head injury: Frequency, duration, and consequences. Arch Surg 2001;136:1118-1123.
25. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;2:81-84.
26. Menegazzi JJ, Davis EA, Sucov AN, et al. Reliability of the Glasgow Coma Scale when used by emergency physicians and paramedics. J Trauma 1993;34:46-48.
27. Fielding K, Rowley G. Reliability of assessments by skilled observers using the Glasgow Coma Scale. Aust J Adv Nurs 1990;7:13-21.
28. Narayan RK, Greenberg RP, Miller JD, et al. Improved confidence of outcome prediction in severe head injury. J Neurosurg 1981;54: 751-762.
29. Marshall LF, Gautille T, Klauber MR, et al. The outcome of severe closed head injury. J Neurosurg (Suppl) 1991;75:28-36.
30. Faul M, Wald MM, Rutland-Brown W, et al. Using a cost-benefit analysis to estimate outcomes of a clinical treatment guideline: Testing the Brain Trauma Foundation guidelines for the treatment of severe traumatic brain injury. J Trauma 2007;63:1271-1278.
31. Cole JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: Effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med 2007;35:568-578.
32. Curley G, Kavanagh BP, Laffey JG. Hypocapnia and the injured brain: More harm than benefit. Crit Care Med 2010;38:1348-1359.
33. Chestnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma 1993;34:216-222.
34. Wakai A, Roberts IG, Schierhout G. Mannitol for acute traumatic brain injury. Cochrane Database Syst Rev 2007;1: CD001049.
35. Kao-Chang L, Chih-Ho C, Wei-Lung C, et al. The early response of mannitol infusion in traumatic brain injury. Acta Neurol Taiwan 2008;17:26-32.
36. Sorani MD, Morabito D, Rosenthal G, et al. Characterizing the dose-response relationship between mannitol and intracranial pressure in traumatic brain injury patients using a high frequency physiologic data collection system. J Neurotrauma 2008;25:291-298.
37. Munar F, Ferrer AM, de Nadal M, et al. Cerebral hemodynamic effects of 7.2% hypertonic saline in patients with head injury and raised intracranial pressure. J Neurotrauma 2000;17:41–51.
38. Kamel H, Navi BB, Nakagawa K, et al. Hypertonic saline versus mannitol for the treatment of elevated intracranial pressure: A meta-analysis of randomized clinical trials. Crit Care Med 2011;39:554-559.
39. Globus MY, Alonso O, Dietrich WD, et al. Glutamate release and free radical production following brain injury: Effects of posttraumatic hypothermia. J Neurochem 1995;65: 1704–1711.
40. Jiang JY, Liang YM, Luo QZ, et al. Effect of mild hypothermia on brain dialysate lactate after fluid percussion brain injury in rodents. Neurosurgery 2004;54:713–717.
41. Lyeth BG, Jiang JY, Robinson SE, et al. Hypothermia blunts acetylcholine increase in CSF of traumatically brain injured rats. Mol Chem Neuropathol 1993;18:247–256.
42. Dietrich WD, Bramlett HM. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics 2010;7:43-50.
43. Fox JL, Vu EN, Doyle-Waters M, et al. Prophylactic hypothermia for traumatic brain injury: A quantitative systemic review. CJEM 2010;12: 355-364.
44. Lowenstein DH. Epilepsy after head injury: An overview. Epilepsia 2009;50(Suppl.2):4-9.
45. Jensens FE. Posttraumatic epilepsy. Treatable epileptogenesis. Epilepsia 2009;50(Suppl.2):1-3.
46. Yablon SA. Postraumatic seizures. Arch Phys Med Rehabil 1993;74: 983-1001.
47. Temkin NR, Dikmen SS Wilensky AJ, et al. A randomized, double blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med 1990;323:497-502.
48. Temkin NR. Preventing and treating posttraumatic seizures. The human experience. Epilepsia 2009;0(Suppl 2):10-13.
49. Temkins NR, Dikmen SS, Anderson GD, et al. Valproate therapy for prevention of post traumatic seizures: A randomized trial. J Neurosurg 1999;91:593-600.
50. Manaka S. Cooperative prospective study on posttraumatic epilepsy: Risk factors and the effects of prophylactic anticonvulsant. Jpn J Psychiatry Neuro 1992;46:311-315.
51. Young B, Rapp RP, Norton JA, et al. Failure of prophylactically administered phenytoin to prevent late posttraumatic seizures. J Neurosurg 1983;58:236-241.
52. Roberts I, Yates D, Sandercock P, et al. Effects of intravenous corticosteroids on death within 14 days in 10.008 adult with clinically significant head injury (MRC CRASH trial): Randomized placebo control trial. Lancet 2004;364:1321-1328.
53. Watson NF, Barber JK, Doherty MJ, et al. Does glucocorticoid administration prevent late seizures after head injury? Epilepsia 2004;45: 690-694.
54. Alderson P, Ian Roberts. Corticosteroids for acute brain injury. The Cochrane Library, July 2009.
55. Eerenberg ES, Kamphuisen PW, Sijpken MK, et al. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: A randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011;124:1573-1579.
The first part of this series dealt with guidelines for the care of ischemic stroke. This issue applies the same principles of evidence-based medicine to hemorrhagic stroke and traumatic brain injury. Hemorrhagic stroke is less common than ischemic stroke, but can be more devastating.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.