Successful Management of Oral Anticoagulation
Authors: Benjamin J. Epstein, PharmD, Postdoctoral Fellow, Departments of Pharmacy Practice and Community Health and Family Medicine, Colleges of Pharmacy and Medicine, University of Florida, Gainesville; and Louis Kuritzky, MD, Professor of Medicine, Department of Community Health and Family Medicine, College of Medicine, University of Florida, Gainesville.
Peer Reviewer: Mark Knudson, MD, MSPH, Vice Chair for Education, Wake Forest University School of Medicine, Winston-Salem, NC.
Appropriate use of anticoagulants offers both opportunity and challenge for the primary care clinician (PCC). Thrombosis, comprised of myocardial infarction (MI) and unstable angina, stroke, pulmonary embolus (PE), and deep venous thrombosis (DVT), is the most common cause of death in the United States. Atrial fibrillation (AF) is the most common arrhythmia requiring hospitalization and is responsible for as much as one-third of strokes. Warfarin is remarkably effective in each of these settings, yet recent data indicate that as few as 35% of persons with AF managed by PCCs receive warfarin, and among those, the international normalized ratio (INR) is in the therapeutic range only half of the time.1 The compelling epidemiologic burden of arterial and venous thrombotic disease is destined to become even more demanding, considering the demographic changes in the U.S. population, which indicate a burgeoning senior citizenry. For instance, AF has its peak incidence between the ages of 75-79,2 an age group that is projected to double by the year 2040.
Anticoagulants in the at-risk population can provide dramatic risk reduction. For instance, meta-analyses of stroke reduction by means of warfarin anticoagulation indicate that AF patients may experience as great as 59% risk reduction in stroke; benefits for secondary prevention of stroke in AF are even greater.3 These same data indicate a mortality reduction of greater than 25%. Given the extraordinary disease burden and remarkable efficacy of anticoagulation, anticoagulants would be more widely embraced and employed if clinicians felt more comfortable with safe use of available agents and confident of benefits to be accrued. This discussion is directed toward simplifying the pathophysiology and effective use of anticoagulation in the primary care setting. — The Editor
Evolving Anticoagulants
Anticoagulants may be grouped broadly under the categories of oral vitamin K antagonists (warfarin [Coumadin]), direct thrombin inhibitors (lepirudin [Refludan], bivalirudin [Angiomax], argatroban [Acova], ximelagatran [Exanta] [available in Europe, but not yet FDA approved in the United States]), and heparinoids (unfractionated heparin [UFH], low molecular weight heparins [LMWHs], pentasaccharides). At this time, the main role for PCCs in the ambulatory setting will be optimal application of warfarin. Warfarin is the only available oral anticoagulant in the United States. Warfarin originally was approved for use in the United States in 1954 and derives its name from the organization that discovered it, the Wisconsin Alumni Research Foundation (WARF). Managing oral anticoagulation in the primary care setting will be the central focus of the discussion to follow.
The Clotting Cascade
A brief overview of the clotting cascade is helpful in understanding the mechanism of action of warfarin. To maintain hemostasis without evoking thomboembolism, there exists a delicate equilibrium between anticoagulant and procoagulant mediators in the blood. Any factors, extrinsic or intrinsic, that disrupt this balance can predispose patients to thrombosis, hemorrhage, or even a combination of the two (e.g., disseminated intravascular coagulation [DIC] or heparin-induced thrombocytopenia syndrome [HITS]). In patients experiencing thromobosis, one or more of the following three factors (referred to as Virchow’s triad) typically are present: venous stasis, hypercoagulability, and/or endothelial injury. Once provoked, the clotting cascade progresses through a series of interrelated reactions, the end result of which is the generation and deposition of fibrin, the activation of platelets, and the formation of a thrombus. (See Figure 1.) The rate-limiting step in this process is activation of factor II (prothrombin) to factor IIa (thrombin). Activated factor II subsequently catalyzes the conversion of fibrinogen to fibrin, which serves as the meshwork for clot formation and propagation.
Mechanism of Action
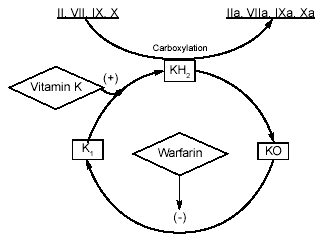
Warfarin often is referred to as a vitamin K antagonist due to its avidity for preventing the activation of the vitamin K-dependent clotting factors (II, VII, IX, and X) to their activated forms (IIa, VIIa, IXa, and Xa). More specifically, warfarin inhibits the cyclic interconversion of vitamin K and its 2,3-epoxide (vitamin K epoxide).4 (See Figure 2.) Normally, vitamin K cycles from a reduced form, vitamin KH2, to an epoxide and back to vitamin K1. Warfarin inhibits the enzyme vitamin K epoxide reductase, thereby reducing the availability of the cofactor (vitamin KH2) necessary for the carboxylation (i.e., activation) of vitamin K-dependent clotting factors. Consequently, factors II, VII, IX, and X circulate in the inactive form, incapable of driving the cascade. In this way, warfarin elicits an anticoagulant response. Because warfarin does not alter the functionality of factors already activated, its onset of action is delayed until those factors are cleared from circulation. Also, note from Figure 1 why administration of active vitamin K is effective for reversal of warfarin-induced anticoagulation: The warfarin-sensitive step is bypassed.
Figure 2. Mechanism of Action of Warfarin
Important Pharmacokinetic Concepts
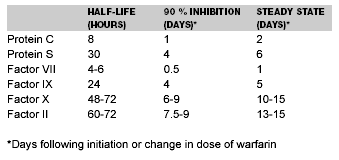
The pharmacokinetics of warfarin often are misunderstood and can result in inappropriate dosing decisions. The following sections will attempt to clarify several important idiosyncrasies specific to warfarin. In contrast to other medications where the half-life of the drug is the determinant of the onset and maintenance of the steady state effect, this is not the case with warfarin. Instead, the half-lives of the clotting factors determine warfarin’s activity since their depletion correlates with the onset and maintenance of anticoagulation. When a patient has been on a stable dose of warfarin for a period of time equal to five times the half-life of the rate-limiting cofactor (factor II), the INR can be considered at steady state because after this time more than 95% of the expected effect of warfarin upon coagulation factors will have occurred. (See Table 1.) Factor II serves as the catalyst for fibrin formation; therefore, its depletion is the most important for gauging the anticoagulant effect of warfarin. Note that factor II has the longest half-life of the vitamin K-dependent clotting factors—this is the reason why several days are required to elicit an adequate anticoagulant response with warfarin. Furthermore, this is the basis for the recommendation that heparin be continued for a minimum of five days in patients experiencing venous thromboembolism (VTE) (i.e., even if the INR is measurably altered by warfarin, full status of anticoagulation as measured by inactivation of factor II, will not be reliably in place until at least five days of warfarin treatment). Based on the half-life of factor II, anticoagulation will approach 90% (three half-lives) of steady state in the first week of therapy in most patients, signaling 90% of the expected effect of warfarin on factor II and a considerable degree of anticoagulation as evidenced by the INR. True steady state will be achieved within two weeks (approximately five half-lives).
Table 1. Vitamin K-Dependent Clotting Factors
Several other concepts regarding warfarin’s mechanism of action are worthy of mention. First, Protein C (PC) and Protein S (PS), the body’s natural anticoagulants, also are inhibited by warfarin. These factors normally help to balance the procoagulant effects of the clotting cascade. Unfortunately, as mentioned previously, warfarin does not suppress factors in the cascade at the same rate. In fact, because the half-life of PC is considerably shorter than that of factor II, PC levels decline before factor II is substantially affected. As a result, initiation of warfarin could promote an initial, transient state of hypercoagulability until factor II levels have been sufficiently suppressed as to balance the decline in PC.5 The authors are unaware of outcome data to confirm the clinical significance of this effect; however, some support may be garnered from the observation of patients with PC deficiency. In this population, there is an increased incidence of warfarin-induced skin necrosis, a rare complication characterized by necrotic lesions due to thrombosis of venules and capillaries in subcutaneous fat that appear three to six days after the initiation of warfarin.4 This suggests that larger initial doses elicit a disproportionately larger decrease in PC vs. factor II in the first week of therapy and that this may be clinically significant in certain populations (e.g., those with low baseline PC levels).
Second, within the first several days after warfarin is administered, the INR response reflects the decline in the factors with the shortest half-life. As a consequence, the INR is a poor surrogate for anticoagulation during the first few days after starting warfarin since it reflects mainly a reduction in factor VII. It is not until sufficient time has elapsed to allow for a significant fall in levels of factor II that the INR accurately reflects the degree of anticoagulation. This takes at least 5-7 days; prior to this, the patient is not completely protected from clot extension. Caution must be observed in the first five days since some patients will achieve an INR above 2 despite inadequate anticoagulation (this can be thought of as a factor VII-INR when the concern really is with a factor II-INR). With these considerations in mind, it is recommended that warfarin be adequately overlapped with heparin for the treatment of thromboembolism. Prior to discontinuing heparin, the patient should receive a minimum of five days of parenteral anticoagulation, and the INR should be therapeutic for 48 hours. The hospital course can be shortened by substituting subcutaneous LMWH, administered at home, for intravenous UFH.
Initiation
In patients presenting with VTE, initial treatment is intended to achieve rapid anticoagulation in an attempt to prevent propagation of the thrombus. Warfarin should be started simultaneously with LMWH or UFH to elicit rapid anticoagulation. A direct thrombin inhibitor (e.g., lepirudin) or synthetic pentasaccharide (e.g., fondaparinux [Arixtra]) are reasonable alternatives depending on the clinical situation (e.g., history of heparin-induced thrombocytopenia). As discussed above, if parenteral antithrombotic therapy is discontinued before five days or the 48-hour overlap threshold, the patient could be at risk for clot extension/embolism since thrombin levels are not sufficiently suppressed by warfarin before that time. Furthermore, discontinuation of heparin may result in a period of rebound hypercoagulability that could further predispose patients to thrombosis.6
There is much debate about the optimum initial dose of warfarin. Historically, a loading dose of 1.5 mg/kg was administered to produce the most rapid effect. This practice eventually was shown to be no more effective than loading doses of 15-20 mg, and smaller doses were less likely to provoke excessive anticoagulation. Today, there is disagreement about whether higher initial doses of 10 mg should be used or whether patients should be started on the average maintenance dose of 5 mg.7 It is not evident from the literature which strategy should be employed. The 10-mg strategy appears to achieve an INR of 2.0 to 3.0 more rapidly (by approximately 1.5 days); however, PC levels decline more precipitously and a larger proportion of patients subsequently are found to have supratherapeutic INRs with this regimen.5,8 The change in protein C levels are less of a concern given that most patients are receiving concomitant therapy with heparin, but overanticoagulation subjects patients to an increased risk of bleeding. It is important to note that a 10-mg loading dose has only been shown safe and effective when used in conjunction with a detailed dosing nomogram and in patients with a low risk for bleeding.9 Without such nomogram, the risk of overanticoagulation and bleeding may be higher compared with a 5-mg strategy. An initial dose of 5 mg daily can be expected to achieve an INR of 2.0-3.0 in 4-5 days in most patients. In consideration of the risks and benefits of both methodologies, and until further data clarify the issues, the authors suggest initiation of therapy with 5 mg daily, unless special circumstances suggest otherwise.
Maintenance
The most critical determinant of the safety and efficacy of anticoagulation with warfarin is the time in the therapeutic range (TTR). The target INR in the majority of patients is 2.5, with an acceptable range of 2.0 to 3.0. (See Table 2.)
Table 2. Intensity of Anticoagulation

There are some data to suggest that patients with mechanical heart valves require a slightly higher INR goal to prevent embolism, 3.0 (2.5-3.5), due to the thrombogenicity of the foreign valve itself. However, the risk of bleeding, including intracerebral hemorrhage, increases in concert with the INR. (See Figure 3.)
Figure 3. Expected Risk of Ischemic Stroke
and Intracranial Bleeding According to INR
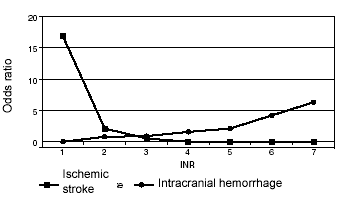
In fact, the relationship becomes nonlinear once the INR exceeds 5.0, with the risk for major bleeding increasing disproportionately more compared to the INR. Conversely, the risk for thromboembolic events rises steeply when the INR is below the therapeutic range. Accordingly, appropriate titration and follow-up are the foundation of successful management of oral anticoagulation. While there is no consensus regarding the exact timing of follow-up, it is evident that the more frequent the monitoring, the greater the TTR.4 When warfarin is started in the hospital, the patient should not be discharged until the INR is therapeutic; however, the dose requirement often changes once the patient returns to a domestic lifestyle. Thus, it is prudent to check the INR in the week after discharge. If the INR is therapeutic, the patient can be seen again in 1-2 weeks for a follow-up INR and counseling; if the INR is also in the therapeutic range at that visit, then monitoring can be extended to every four weeks. If warfarin is initiated in the outpatient setting, the INR should be monitored once or twice per week until therapeutic, then every two weeks until the INR is stable and in the therapeutic range during two consecutive visits, at which time monitoring can be performed every four weeks. Four weeks is the maximum recommended interval between visits in stable patients. More frequent monitoring will increase the TTR, but becomes overly cumbersome to be practicable for most patients. The authors’ anticoagulation clinic employs monthly monitoring for patients at goal and without compelling reason for more frequent follow-up (e.g., new interacting medication or acute illness). Patients with supratherapeutic or subtherapeutic INRs are monitored more frequently depending on the clinical situation. (See below.)
The relationship between warfarin dose and INR is nonlinear; thus, small dose adjustments should be made to avoid overcorrection. The dose should be adjusted up or down in increments of 10-15 % per week if the patient’s INR is not at goal. For example, in a relatively stable patient on 5 mg every day who presents to clinic with an INR of 1.8 (goal 2.0-3.0) and no bleeding for two consecutive visits, the total weekly dose could be increased to 5 mg (1 tablet) every day except 7.5 mg (1½ tablets) on Tuesday and Thursday and a follow-up INR obtained in 1-2 weeks. Altering the timing and spacing of dosage is based upon common sense and basic pharmacokinetic principles. For instance, take the case of a patient presenting to clinic on Monday with a supratherapeutic INR who is taking warfarin 5 mg every day except Wednesday and Saturday, on which he/she takes 7.5 mg each day. The clinician should reduce or omit the first possible dose, which will tend to reduce the INR, rather than waiting until later in the week. It often is helpful to look for a trend in the INR. For instance, if a given patient’s INR increases from 1.7 to 2.4 to 3.0 at weekly intervals, the clinician should try to identify an underlying cause, determine if the dose is too high, and see the patient back before four weeks (i.e., in 1-2 weeks even though the INR is in range for two consecutive weeks).
Monitoring should be accelerated following dose changes to resemble that during the initial titration and then extended to four-week intervals once the INR is in the therapeutic range and at steady state. In addition, there often are instances when changes in the INR are predictable, such as when a patient is started on a new medication that interacts with warfarin (e.g., metronidazole [Flagyl]) or the consumption of vitamin K is expected to change. Under these circumstances it is prudent to monitor the INR more frequently (e.g., weekly). Also, a preemptive dose adjustment may be warranted. For example, in a patient started on amiodarone [Cordarone, Pacerone], it is recommended that the maintenance dose of warfarin be decreased empirically by 25-50% depending on the dose of amiodarone.
Duration of Therapy
The duration of therapy is dependent upon the indication for oral anticoagulant therapy and the perceived risk of bleeding and recurrent thromboembolism. The philosophy regarding appropriate duration of anticoagulation is in a state of evolution. Although it is convenient to divide thromboembolism patients into those with a precipitating event (e.g., surgery, bedridden status, long-haul flight, oral contraceptives) vs. idiopathic, this classification ignores the fact that even though a recent precipitant may have been present, the huge majority of patients who experience that precipitant do not experience a thromboembolic event. In other words, of the millions of patients who engage in long-haul flights, only a handful of patients experience DVT, and hence, one must wonder if there is not something thrombophilic at work even in these patients who have an apparent trigger for their event. This observation, coupled with long-term data in unselected DVT patients that confirms risk reduction even in the setting of very long term prophylaxis (2-4 years), has prompted clinicians to re-think the optimum duration of anticoagulation. Patients with AF generally require indefinite treatment. Two notable exceptions are patients with lone AF (i.e., AF with no risk factors for stroke, which include prior ischemic stroke, TIA, or systemic embolism, age older than 75 years, moderately or severely impaired left ventricular systolic function and/or congestive heart failure, history of hypertension, or diabetes mellitus) or new-onset AF that terminates spontaneously. Patients with lone AF should be treated with 325 mg of aspirin daily since aspirin and warfarin are similarly effective in this population and aspirin is associated with less bleeding; however, these patients are the exception because the majority of patients with AF have at least one other risk factor for stroke. In patients with new-onset AF after cardiothoracic surgery, for example, AF often terminates spontaneously or can be electrically or chemically cardioverted. In this population, the risk for recurrence is relatively low and an indication for life-long oral anticoagulation may be unnecessary. Patients who do not satisfy either of these exceptions should be anticoagulated indefinitely. This includes most patients with AF who are managed with rhythm control even if the patient is in normal sinus rhythm during office visits. Antiarrhythmic therapy to maintain normal sinus rhythm is inherently unsuccessful and the risk of thromboembolism may persist despite apparently successful rhythm control.
Table 3 provides guidelines for the duration of therapy in patients with DVT or PE. In patients experiencing an initial VTE associated with an identifiable risk factor, management is controversial, but treatment should be given for at least three months since a shorter duration of therapy is associated with an unacceptable risk of recurrence.10 The decision to treat longer should be based on the patient’s risk of bleeding vs. the risk of recurrence. If the risk factor is unavoidable (e.g., cancer), treatment should continue as long as the risk factor is present. For a transient risk factor (e.g., immobilization), the decision to discontinue therapy must consider the risk of a recurrent event. The rationale for extended treatment is based upon the deductive premise that because many patients are exposed to transient risk factors (e.g., long-haul airplane flights, oral contraceptives, etc.) and very few develop VTE, those that do likely have an underlying predisposition to VTE.
Table 3.
Duration of Oral Anticoagulation
for Common Indications

A major change in the most recent consensus guidelines on antithrombosis is that in patients suffering from a first idiopathic VTE, oral anticoagulation should be considered indefinitely.11 In this setting, it is critical to periodically weigh the risk:benefit ratio and discuss treatment options with the patient. It is clear that 6-12 months should be considered the minimum duration of therapy, considering that the risk of recurrence in the first year after discontinuation is as high as 27% when treatment is discontinued at three months and 10% at six months. There is convincing evidence that longer periods of therapy (as long as four years) are superior to shorter periods (six months) in patients with recurrent VTE.12 As a result, physicians tend to treat patients longer provided the patient is a good candidate for anticoagulation (i.e., no bleeding diathesis, especially gastrointestinal; no substance or alcohol abuse; and capable of adherence). Since low-intensity anticoagulation (INR goal of 1.5-2.0) has been shown to be less effective for prevention of VTE than a goal of 2.0-3.0 with no reduction in risk of bleeding, there is no role for low-intensity anticoagulation.13
Dealing with Supratherapeutic INRs
Clinicians frequently are confronted with the dilemma of overanticoagulation, which may be due to co-morbid illness, drug-drug interactions, dietary changes, or for no apparent reason. It is clear that these excursions significantly increase a patient’s risk for clinically important bleeding.14,15 Determination of the patient’s bleeding risk and ruling out active hemorrhage are critical next steps. (See Figure 4.) Given that intensity of anticoagulation is the most reliable predictor of hemorrhage, the goal of treating patients with supratherapeutic INRs is to rapidly restore the INR to within the therapeutic range while minimizing the risk of thromboembolism. High doses of vitamin K are effective but may lower the INR more than is necessary and can provoke warfarin resistance (excess vitamin K is stored in the liver and decreases the effectiveness of warfarin) for a week or more, which of course places the patient at risk of recurrent thromboembolism. Any patient who presents with major bleeding should be admitted promptly and administered plasma and parenteral vitamin K. For patients without major bleeding, the simplest and most widely implemented approach is to withhold warfarin and allow the INR to fall into the therapeutic range.16 The INR will fall approximately 1-2 units each day. Keep in mind, however, that if the INR was increasing at the time warfarin was discontinued, it may continue to increase or fail to change for an additional 24 hours before decreasing. Several case series have validated that in patients with an INR of 6.0-10.0, the risk of hemorrhage is minimal (approximately 0.6%) with this approach.17,18 Nevertheless, without adequately controlled clinical trials, it is prudent to stratify patients according to bleeding risk, treating those at highest risk more aggressively. (See Table 4.) For example, an INR of 8 in a 76-year-old female with AF, chronic renal insufficiency, and history of gastrointestinal bleeding who is on concomitant antiplatelet therapy should be treated more aggressively (e.g., withhold warfarin and administer 2.5 mg oral phytonadione) than the same INR in a 52-year-old-male with AF and no other risk factors (e.g., withhold warfarin and recheck INR in 2-3 days). When vitamin K is indicated, the oral route should be used in most cases. Results of three prospective studies show that withholding warfarin and administering 1.0 mg of vitamin K results in an average decrease in the INR from 5.7 to 2.8 on the day after vitamin K is administered.16,19 Based on these results, microdose vitamin K (i.e., less than 5 mg) should be the preferred strategy in most situations when vitamin K is indicated since it lowers the INR rapidly, does not predispose patients to anaphylactoid or skin reactions, and is unlikely to induce warfarin resistance. Figure 4 summarizes treatment strategies for managing supratherapeutic INRs.
Table 4. Factors Identified as Risk Factors
for
Increased Risk of Bleeding
on Anticoagulation

Periprocedural Management
Anticoagulation in patients who undergo surgical procedures can be approached in one of two ways: an aggressive strategy that substitutes either full doses or prophylactic doses of UFH or LMWH for warfarin before and after the procedure, or a minimalist strategy, in which all anticoagulation is discontinued before the procedure and no heparin is administered after the procedure. The appropriate strategy should be selected based on the patient’s risk of thrombosis and bleeding.20 Keep in mind that the risk for bleeding usually is overestimated and the risk for thrombosis underestimated. This observation is crystallized by the perception of the risk of bleeding during dental procedures. Many physicians prefer to discontinue anticoagulation prior to such procedures despite the fact that thrombosis is more likely to occur if warfarin is stopped than is major bleeding if warfarin is continued. Dental colleagues often request physician consultation to recommend management of anticoagulation in the perioperative period for dental work. Consensus suggests that intensive local maneuvers, rather than modulation of anticoagulation, generally are sufficient for all but the most invasive dental procedures.21,22
If a minimalist strategy is selected, discontinuing warfarin will result in an INR of less than 1.5, a degree of anticoagulation considered safe for most surgical procedures, in four days in the majority of patients with an INR in the therapeutic range prior to withholding warfarin. Keep in mind, however, that the early decrease in the INR is a function of increasing factor VII levels rather than factor II, and excessive bleeding could occur despite a normal INR (because factor II activity still is suppressed). With the minimalist approach of withholding warfarin four days before surgery and restarting it as soon as possible after surgery, patients will have a subtherapeutic INR for approximately two days before surgery and two days after surgery. This strategy is appropriate for patients with a low risk of thromboembolism. For instance, in patients with nonvalvular AF who have not had a systemic embolism, the annual incidence of such an event is approximately 4.5% per year without anticoagulation.23 Hence, the risk of four days without full anticoagulation is expected to be minimal and warfarin can be restarted immediately after the procedure.
If an aggressive strategy is being considered, there are several important points to keep in mind. In patients at risk of arterial thromboembolism, there is a stronger argument in favor of preoperative heparin since the risk of thromboembolism is similar before and after surgery, while the risk of bleeding is amplified after surgery. In patients at high risk for VTE, the risk of a recurrent VTE is magnified after surgery, and the case for postoperative anticoagulation is stronger. The magnitude of the increase in risk varies with the time since the previous VTE. In the first month after a venous event, the risk of recurrence is sufficiently high that therapeutic doses of heparin are justified preoperatively and postoperatively. In the second and third month after a VTE, the risk of VTE continues to outweigh the risk of bleeding with postoperative heparin since surgery increases the risk of VTE 100-fold.20 Preoperative heparin is justified in this population if major risk factors for VTE are present. Beyond the third month, the risk of bleeding usually outweighs the risk of recurrent VTE, and strategies other than therapeutic doses of heparin (e.g., prophylactic doses of UFH or LMWH or intermittent pneumatic compression) should be employed to reduce the risk of VTE. For more specific recommendations regarding the periprocedural management of anticoagulation, the reader is referred to the recent American College of Chest Physicians (ACCP) guidelines.4
Other Considerations
An overview of all interactions with oral anticoagulation is outside the scope of this paper. Clinicians are encouraged to refer to more complete references for this purpose.4,24 Instead, the following section reviews different scenarios that are encountered frequently.
Aspirin. It is important to differentiate between the antiplatelet effect of aspirin and the anticoagulant effect. Aspirin irreversibly inhibits platelet function when administered in doses as low as 30 mg per day. By inhibiting platelet function, aspirin may increase the risk for bleeding in patients that are anticoagulated. In the Warfarin, Aspirin, Reinfarction Study (WARIS), the annual risk of nonfatal major bleeding increased from 2.14% to 2.70% when aspirin was added to oral anticoagulation with warfarin.25 Of importance is the effect of aspirin on prostaglandins in the gastrointestinal mucosa. Even low doses of aspirin increase the risk for gastrointestinal toxicity, and these events are potentiated by concomitant use of oral anticoagulants. At higher doses (more than 1.5 g/d), aspirin also suppresses coagulation through an anti-vitamin K effect. When used at these doses for analgesia or antipyresis, aspirin can prolong the INR and increase the risk for hemorrhage.
Despite these risks, aspirin continues to have an important role in select patients on warfarin. Low-dose aspirin (75-100 mg) is indicated in conjunction with warfarin in patients with caged ball or disk heart valves as well as individuals who have undergone valve replacement and sustain a systemic embolic event despite adequate oral anticoagulation. In these populations, the risk of future embolic events outweighs the incremental bleeding risk. The role of aspirin in patients on warfarin who have coronary artery disease (CAD) and a separate indication for anticoagulation (e.g., AF) continues to be a matter of debate. In the past, warfarin was believed to obviate the need for antiplatelet therapy; however, elucidation of the pathophysiology underlying venous and arterial thrombotic events has called that rationale into question and spawned several clinical trials exploring this matter.26-30 A meta analysis of randomized trials that included data from more than 20,000 patients with established CAD recently addressed the issue.31 Based on the results of this analysis, several conclusions can be made. First, the combination of low-intensity anticoagulation with warfarin (INR less than 2.0) and aspirin confers no benefit over aspirin alone in patients with CAD. It does, however, subject the patient to a considerable increase in major bleeding episodes. Second, when moderate- to high-intensity anticoagulation administered in conjunction with low-dose aspirin was compared with warfarin alone, recurrent ischemic events were reduced by 14%, but the result was not statistically significant. However, there is probably a subpopulation of patients at high risk of coronary events (e.g., diabetes, hyperlipidemia, or hypertension) in whom the addition of low-dose aspirin would confer a benefit that outweighs the small increase in bleeding. Regardless, it is prudent to monitor the INR more carefully in patients on concomitant antiplatelet and anticoagulant treatment.
Nonsteroidal Anti-inflammatory Drugs (NSAIDs) and COX-2 Inhibitors. The greatest concern with concomitant use of NSAIDs is the considerable risk of ulcerations, bleeding, or perforations, the consequences of which can be devastating in patients receiving anticoagulation. For this reason, it is prudent to avoid NSAIDs in this population. Cyclooxygenase (COX)-2 selective inhibitors emerged as a viable alternative in patients at significant risk of gastrointestinal bleeding. However, the recent withdrawal of rofecoxib (Vioxx) and the lack of gastroprotection in patients on low-dose cardioprotective aspirin, has called into question the safety of the class. The increased risk appears to be a class effect. A randomized, controlled trial with lumiracoxib (not yet approved) showed harm,32 as did a meta-analysis with valdecoxib (Bextra), and a clinical trial with celecoxib (Celebrex) was recently terminated due to cardiovascular events. There also is a biologically plausible mechanism to suggest that this is a class effect. Because prostacyclin production is mediated by COX-2 and thromboxane production is mediated by COX-1, drugs that are selective for COX-2 will create a mismatch between vascular thromboxane and prostacyclin, which may lead to a prothrombotic state.33 One might consider the risk as a spectrum, with more selective drugs conferring more risk and less selective drugs less risk. An alternative strategy in patients at risk of cardiovascular events who have an indication for anti-inflammatory medications but require gastroprotection is the addition of a proton pump inhibitor (PPI) to a nonselective NSAID, which reduces the bleeding risk to a similar extent as administering a COX-2 selective agent alone (i.e., the risk is reduced, though not completely).34
Acetaminophen. Acetaminophen is recognized as the preferred analgesic in patients on concomitant anticoagulants because it is devoid of antiplatelet activity and does not interfere with the integrity of the gastrointestinal mucosa. As a result, there is a misconception that acetaminophen does not interact with warfarin. In reality, there is a strong, dose-dependent relationship between acetaminophen ingestion and excessive anticoagulation.35 The mechanism behind this often overlooked interaction has not been fully elucidated, but may be related to effects on cytochrome P450 isozymes or interconversion of vitamin K-dependent clotting factors. As a consequence, patients who use acetaminophen in the primary care setting must be monitored more closely for anticoagulant instability, especially when acetaminophen is used periodically. Patients on a stable dose of acetaminophen should be at lower risk since the interaction between acetaminophen and warfarin will stabilize.
Antibiotics. Any antibiotic can interfere with the enterohepatic recirculation of vitamin K by disrupting the enteric flora, thereby decreasing the availability of vitamin K and increasing the INR. Likewise, infection itself increases metabolic demand and can accelerate the degradation of vitamin K-dependent clotting factors. With less vitamin K available, fewer clotting factors can be activated. Metronidazole and trimethoprim/sulfamethoxazole (Bactrim, Septra) inhibit cytochrome P450 2C9, which is responsible for the metabolism of the more active S-isomer of warfarin, potentiating warfarin’s effect on the INR. Second- and third-generation cephalosporins augment the INR via a direct effect on the cyclic interconversion of vitamin K. On the other hand, dicloxacillin (Dynapen) and rifampin (Rifadin) can induce the metabolism of warfarin and decrease its effectiveness.
Thyroid Replacement. Exogenous administration of thyroid hormones can result in fluctuations in the INR. The distinction between thyroid replacement and other drugs that induce metabolism is an important one. Thyroid replacement therapy increases metabolism, but not of warfarin (like some other drugs); instead, the rate at which clotting factors are metabolized is accelerated. This results in an increase in the INR and decreased dose requirement in patients who are initiated on thyroid replacement therapy or have their dose increased. The reverse is true when replacement therapy is discontinued or the dose is decreased.
Diet. One of the many limitations of vitamin K antagonists is the numerous interactions with foods that contain vitamin K.36 Patients often misunderstand the counseling they receive regarding this interaction. Many perceive that vitamin K-containing foods must be avoided entirely. In reality, consistency should be stressed to the patient so that he or she does not feel compelled to eliminate these types of foods from the diet. Patients can be counseled to eat a specific number of servings of vitamin-K-containing foods each week or each day. Physicians also should recognize the therapeutic potential of these foods during treatment. Instead of changing the dose of warfarin, omitting or adding one to two servings of vegetables per week may be sufficient to influence the INR slightly upward or downward, respectively, obviating the need to adjust the dose. This interaction requires persistence on the part of the physician to ensure that the patient is fully cognizant of the effects of vitamin K on the INR. In patients whose INR will not stabilize due to dietary indiscretion, some clinicians will prescribe a vitamin K supplement 100-200 mcg daily to dampen the fluctuating intake in the diet. This minimizes peaks and troughs in intake by setting a higher baseline. If this strategy is employed, the authors encourage physicians to initiate vitamin K only when the INR is near the upper portion of the therapeutic range and increase the frequency of monitoring accordingly.
Point of Care Testing
The widespread availability of point of care (POC) INR testing has streamlined the management of oral anticoagulation in the professional setting and for patient self-management (PSM) or self-testing (PST). POC devices usually analyze a fingerstick sample of capillary blood and employ a thromboplastin-mediated clotting time. The device converts the clotting time to a prothrombin time and expresses the value as an INR. For the most part, the accuracy of POC devices has been adequately validated. Limitations include larger disparities in reported INR compared to standard plasma-based assays, especially as the INR increases above the therapeutic range; errors when calibrating the international sensitivity index (ISI) of the device; the inability to calculate a mean normal PT; and significant cross-reactivity with antibodies in patients with antiphospholipid antibody syndrome (APS). These differences may lead to different dosing decisions in clinical practice. Unfortunately, the limitations are irreconcilable with current monitors; newer generation devices will need to incorporate different ISI calibration methods to overcome these shortcomings. Of note, POC devices are not indicated for use in patients with APS since the thromboplastin used in these devices cross-reacts with antiphospholipid antibodies, producing an unreliable result.
There is insufficient evidence in the medical literature to make definitive recommendations regarding the role of PSM or PST. The available evidence suggests that PSM or PST might result in more INRs in the therapeutic range compared to usual care (dose management in these models were made by an anticoagulation monitoring service [AMS]). On the other hand, PSM and PST achieve similar TTR compared with AMS. Unfortunately, these studies did not control the level of patient education, compliance, frequency of monitoring, and variability in the reagents and testing devices used, making the results difficult to apply in clinical practice. One alternate explanation for improved control is that patients testing at home might test more often, which has been shown to increase the TTR, or that dose adjustments made by the AMS were more effective compared to usual care. Recently, PSM was shown to result in a similar level of INR control and fewer complications compared to management by hematologists in an anticoagulation clinic.37
Conclusion
Warfarin remains a mainstay for patients that require long-term anticoagulation. While it appears likely that less cumbersome alternatives to warfarin are on the horizon, current setbacks in development have delayed their arrival. In the meantime, physicians must be proficient in the management of patients on warfarin. This can be difficult given the lack of a unifying standard for initiation, titration, monitoring, and follow-up. Furthermore, several issues regarding management are controversial; this article has attempted to address several of those issues here. The plethora of clinical trial data, which continues to emerge, and inconsistencies in the literature can make for uncertainty when it comes to treating patients in clinical practice. This article has summarized the important aspects of managing oral anticoagulants based upon current guidelines, the clinical trial data, and the authors’ own experiences to provide a practical reference for clinicians treating patients in the ambulatory setting.
References
1. Samsa GP, Matchar DB, Goldstein LB, et al. Quality of anticoagulation management among patients with atrial fibrillation: Results of a review of medical records from 2 communities. Arch Intern Med 2000;160:967-973.
2. Feinberg WM, Blackshear JL, Laupacis A, et al. Prevalence, age distribution, and gender of patients with atrial fibrillation. Arch Intern Med 1995;155:469-473.
3. Hart RG, Benavente O, McBride R, et al. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: A meta-analysis. Ann Intern Med 1999;131:492-501.
4. Ansell J, Hirsh J, Poller L, et al. The pharmacology and management of the vitamin K antagonists: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126: 204S-233S.
5. Weiss P, Soff GA, Halkin H, et al. Decline of proteins C and S and factors II, VII, IX and X during the initiation of warfarin therapy. Thromb Res 1987;45:783-790.
6. Bijsterveld NR, Peters RJ, Murphy SA, et al. Recurrent cardiac ischemic events early after discontinuation of short-term heparin treatment in acute coronary syndromes: Results from the Thrombolysis in Myocardial Infarction (TIMI) 11B and Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events (ESSENCE) studies. J Am Coll Cardiol 2003;42:2083-2089.
7. Eckhoff CD, Didomenico RJ, Shapiro NL. Initiating warfarin therapy: 5 mg versus 10 mg (December). Ann Pharmacother 2004;38: 2115-2121.
8. Harrison L, Johnston M, Massicotte MP, et al. Comparison of 5-mg and 10-mg loading doses in initiation of warfarin therapy. Ann Intern Med 1997;126:133-136.
9. Kovacs MJ, Rodger M, Anderson DR, et al. Comparison of 10-mg and 5-mg warfarin initiation nomograms together with low-molecular weight heparin for outpatient treatment of acute venous thromboembolism. Ann Intern Med 2003;138:714-719.
10. Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004;2:743-749.
11. Buller HR, Agnelli G, Hull RD, et al. Antithrombotic therapy for venous thromboembolic disease: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004;126(3 Suppl):401S-428S.
12. Ridker PM, Goldhaber SZ, Danielson E, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N Engl J Med 2003;348:1425-1434.
13. Kearon C, Ginsberg JS, Kovacs MJ, et al. Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003;349:631-639.
14. Fihn SD, Callahan CM, Martin DC, et al. The risk for and severity of bleeding complications in elderly patients treated with warfarin. The National Consortium of Anticoagulation Clinics. Ann Intern Med 1996;124:970-979.
15. Hylek EM, Chang YC, Skates SJ, et al. Prospective study of the outcomes of ambulatory patients with excessive warfarin anticoagulation. Arch Intern Med 2000;160:1612-1617.
16. Ginsberg JA, Crowther MA, White RH, et al. Anticoagulation therapy. Hematology (Am Soc Hematol Educ Program) 2001:339-357.
17. Glover JJ, Morrill GB. Conservative treatment of overanticoagulated patients. Chest 1995;108:987-990.
18. Lousberg TR, Witt DM, Beall DG, et al. Evaluation of excessive anticoagulation in a group model health maintenance organization. Arch Intern Med 1998;158:528-534.
19. Crowther MA, Julian J, McCarty D, et al. Treatment of warfarin-associated coagulopathy with oral vitamin K: A randomised controlled trial. Lancet 2000;356:1551-1553.
20. Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997;336:1506-1511.
21. Wahl MJ, Howell J. Altering anticoagulation therapy: A survey of physicians. J Am Dent Assoc 1996;127:625-626, 629-630.
22. Wahl MJ. Dental surgery in anticoagulated patients. Arch Intern Med 1998;158:1610-1616.
23. Aronow WS, Ahn C, Kronzon I, et al. Incidence of new thromboembolic stroke in persons 62 years and older with chronic atrial fibrillation treated with warfarin vs. aspirin. J Am Geriatr Soc 1999; 47:366-368.
24. Cropp JS, Bussey HI. A review of enzyme induction of warfarin metabolism with recommendations for patient management. Pharmacotherapy 1997;17:917-928.
25. Hurlen M, Abdelnoor M, Smith P, et al. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002;347:969-974.
26. Hurlen M, Smith P, Arnesen H. Effects of warfarin, aspirin and the two combined, on mortality and thromboembolic morbidity after myocardial infarction. The WARIS-II (Warfarin-Aspirin Reinfarction Study) design. Scand Cardiovasc J 2000;34:168-171.
27. Cohen M, Adams C, Hawkins L, et al. Usefulness of antithrombotic therapy in resting angina pectoris or non-Q-wave myocardial infarction in preventing death and myocardial infarction (a pilot study from the Antithrombotic Therapy in Acute Coronary Syndromes Study Group). Am J Cardiol 1990;66:1287-1292.
28. Huynh T, Theroux P, Bogaty P, et al. Aspirin, warfarin, or the combination for secondary prevention of coronary events in patients with acute coronary syndromes and prior coronary artery bypass surgery. Circulation 2001;103:3069-3074.
29. The OASIS Investigators. Effects of long-term, moderate-intensity oral anticoagulation in addition to aspirin in unstable angina. J Am Coll Cardiol 2001;37:475-485.
30. Fiore LD, Ezekowitz MD, Brophy MT, et al. Department of Veterans Affairs Cooperative Studies Program Clinical Trial comparing combined warfarin and aspirin with aspirin alone in survivors of acute myocardial infarction: Primary results of the CHAMP study. Circulation 2002;105:557-563.
31. Anand SS, Yusuf S. Oral anticoagulants in patients with coronary artery disease. J Am Coll Cardiol 2003;41:62S-69S.
32. Farkouh ME, Kirshner H, Harrington RA, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: Randomised controlled trial. Lancet 2004;364: 675-684.
33. Howard PA, Delafontaine P. Nonsteroidal anti-inflammatory drugs and cardiovascular risk. J Am Coll Cardiol 2004;43: 519-525.
34. Chan FKL, Hung LCT, Suen BY, et al. Celecoxib versus diclofenac and omeprazole in reducing the risk of recurrent ulcer bleeding in patients with arthritis. N Engl J Med 2002;347:2104-2110.
35. Hylek EM, Heiman H, Skates SJ, et al. Acetaminophen and other risk factors for excessive warfarin anticoagulation. JAMA 1998;279: 657-662.
36. Wells PS, Holbrook AM, Crowther NR, et al. Interactions of warfarin with drugs and food. Ann Intern Med 1994;121: 676-683.
37. Menendez-Jandula B, Souto K, Oliver A, et al. Comparing self-management of oral anticoagulant therapy with clinic management: A randomized trial. Ann Intern Med 2005;142:1-10.
Appropriate use of anticoagulants offers both opportunity and challenge for the primary care clinician. This discussion is directed toward simplifying the pathophysiology and effective use of anticoagulation in the primary care setting.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.