Toxic Alcohols
Toxic Alcohols
Author: Richard J. Church, MD, Fellow, Medical Toxicology, University of Massachusetts Medical and Health Center, Worcester, MA.
Peer Reviewers: Frank LoVecchio, DO, MPH, Research Director, Maricopa Medical Center, Department of Emergency Medicine, Director, Banner Poison Center, Phoenix, AZ; and Steven M. Winograd, MD, FACEP, Attending, Emergency Medicine, Horton Hospital, Middletown, NY, Arden Hill Hospital, Goshen, NY.
Definition and Relevance of the Problem
Exposures to toxic alcohols such as methanol, ethylene glycol, and isopropanol have been reported in the medical literature for decades. These agents are found in a variety of household products, leading to accidental ingestion in the pediatric population and intentional ingestion in the adult population as a suicidal agent or as an inexpensive substitute for ethanol. Poisonings with these agents can result in significant morbidity and mortality and require a heightened awareness on the part of the emergency physician to recognize and manage these exposures.
Methanol
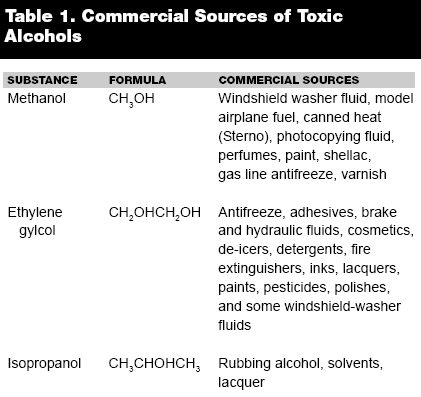
History and Epidemiology. Methanol, or methyl alcohol, was first isolated from distilled boxwood by Robert Boyle in 1661, its molecular composition being determined in 1834 by Dumas and Peligot. Its name stems from the term methylene from the Greek roots for "wood wine." It also goes by the names "colonial spirit," "wood alcohol," and "solvent alcohol." Methanol is a clear, colorless, volatile, highly flammable liquid that is soluble in water and has a musty, fruity, alcoholic taste and smells slightly sweeter than ethanol.1 Its industrial production began in 1923 with over one billion gallons produced annually, and it is used widely as a solvent and in the production of formaldehyde and methylated compounds.2 Methanol-containing consumer products include windshield washer fluid, model airplane fuel, canned heat (Sterno), photocopying fluid, perfumes, paint, shellac, gas line antifreeze, and as an adulterant in "denatured" alcohol. Small amounts of methanol are also found in foods such as fresh fruit, juices, vegetables, and, because it is a natural fermentation product, as a minor ingredient in all spirits.3 (See Table 1.)
 |
The American Association of Poison Control Centers Toxic Exposure Surveillance System reports approximately 2200 methanol exposures per year, with one death for 180 exposures.2 Most poisonings follow ingestions, whether accidental or intentional; however, rare cases have been reported after dermal4 and inhalational exposures.5 Unintentional exposures usually involve consumption of household products by children. Purposeful ingestions involve either ingestion of a commercially available product in a suicide attempt or the consumption of methanol containing products by alcoholics as a cheap substitute for ethanol. Several epidemics of methanol poisoning have been reported in the United States, the most recent involving the ingestion of tainted fermented beverages by 44 inmates at the State Prison in Southern Michigan.6 Although domestic production of moonshine has declined over the past 30 years, global epidemics continue to ravage countries such as Kenya, El Salvador, and Bangladesh.7
Toxicokinetics and Toxicodynamics. Methanol is readily absorbed via gastrointestinal, percutaneous, and inhalational routes, with peak concentrations reached within 30-60 minutes.8,9 After uptake and distribution in the total body water compartment (0.6 L/kg), 97% is cleared from the body by metabolism in the liver; the rest is excreted as unmetabolized methanol in the urine or in expired air.10 Metabolism occurs sequentially, with alcohol dehydrogenase acting as the primary enzyme responsible for the oxidation of methanol to formaldehyde.11 Aldehyde dehydrogenase then rapidly converts formaldehyde to formic acid with no appreciable accumulation of formaldehyde in the blood.12 In the final metabolic step, tetrahydrofolate-dependent reactions transform formic acid to carbon dioxide and water.13 (See Figure 1.) This process is slow and observes Michaelis-Menton kinetics. At low serum concentrations methanol disposition obeys first-order kinetics with a half-life of three hours or less. However, at higher concentrations, such as those seen in overdose, alcohol dehydrogenase is saturated, and elimination becomes a zero-order process with a rate of 8.5 mg/dL/hour.14 Additionally, the limited folate reserve found in primates becomes overwhelmed causing formic acid accumulation, the entity thought to be responsible for the metabolic acidosis in the early stage of methanol poisoning.15
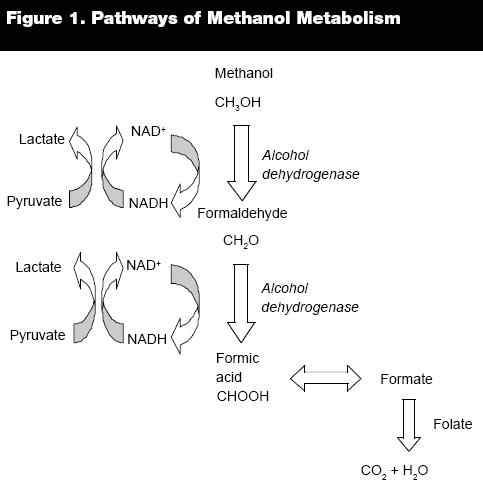 |
Pathophysiology. Signs and symptoms of methanol poisoning can be delayed 12-24 hours as formic acid slowly accumulates, although patients may become symptomatic as early as 2-5 hours after ingestion, implying a dose-dependent response.9 In contrast, latency periods can be as long as 96 hours if patients co-ingest ethanol, a competitive inhibitor of methanol metabolism.16 Once accumulated, formic acid inhibits cytochrome oxidase c activity, leading to tissue hypoxia, lactate formation, and worsening acidosis.11,17 As acidosis worsens, formic acid diffuses more easily across cell membranes (including the blood brain barrier) causing further central nervous system depression, hypotension, and continued increases in lactate production.12 The undissociated formic acid preferentially targets the optic disc and optic nerve causing localized cellular hypoxia and leading to disc edema, myelin sheath breakdown, and optic nerve atrophy.18 The putamen, caudate nucleus, and peripheral white matter are also susceptible to injury, leading to edema, necrosis, and hemorrhage.19
Clinical Manifestations. The clinical features of methanol poisoning involve the triad of severe anion gap metabolic acidosis, visual changes, and mental status depression. The early stage of methanol poisoning may involve mild CNS depression, the mechanism for which is not yet fully elucidated, but presumed to be analogous to the inebriation seen in ethanol consumption, which is mediated through increased GABAergic tone.20,21 Patients are otherwise relatively asymptomatic during this initial period, after which an uncompensated anion gap metabolic acidosis develops. This intermediate stage is manifested by the development of a wide range of visual disturbances from blurred vision to visual field deficits to complete blindness.22 Often, patients will report the "feeling of being in a snow field." The physical examination is notable for central scotoma of visual field testing, hyperemia, and pallor of the optic disc and papilledema.19
Other symptoms associated with methanol intoxication include headache, lightheadedness, abdominal pain, nausea, vomiting, and dyspnea. Methanol exerts a direct toxic effect on the pancreas to produce pancreatitis.23 Acute renal failure, a rare complication of methanol poisoning, is a consequence of myoglobinuria.24 Without treatment, coma and respiratory and circulatory failure may ensue.
Survivors of severe methanol poisoning who have suffered putamenal and subcortical white matter lesions may develop a Parkinson-like extrapyramidal syndrome characterized by rigidity, bradykinesia, mild tremor, masked faces, lethargy, and mild dementia.25
Ethylene Glycol
History and Epidemiology. Ethylene glycol was first synthesized in 1859 by Charles Wurtz and because of its low freezing point (~13°C) and high boiling point (198°C), it started being used as an engine coolant as early as World War II. Although its primary use remains as automotive antifreeze, ethylene glycol can also be found in adhesives, brake and hydraulic fluids, cosmetics, de-icers, detergents, fire extinguishers, inks, lacquers, paints, pesticides, polishes, and some windshield-washer fluids. (See Table 1.) The ingestion of ethylene glycol, intentional or unintentional, results in approximately 5000 poisonings each year, with about 30-40 fatalities.26 Poisonings occur in intentional ingestions as a suicide agent and, because of its sweet taste and intoxicating properties, as an inexpensive substitute for ethanol.27 The vast majority of exposures are through accidental misuse; however, purposeful ingestions account for most documented fatalities.28
Toxicokinetics and Toxicodynamics. Ethylene glycol is absorbed rapidly in the gastrointestinal tract; however, unlike methanol, its percutaneous and pulmonary absorption is very limited due to low volatility and poor skin penetration.29 It is highly water soluble and distributes evenly throughout tissues of the body with a relatively small volume of distribution (0.5-0.8 L/kg).30 Peak concentrations are reached one to four hours after ingestion; approximately 80% of the absorbed dose undergoes hepatic metabolism. The other 20% is excreted unchanged in the urine by the kidneys, the only toxic alcohol to exhibit this effect.31 The elimination half-life of ethylene glycol is approximately 3 hours when following first order kinetics, but increases to 8.5 hours when serum concentrations are higher, such as in overdose.32 Additionally, metabolism is slowed and elimination can be prolonged up to 18 hours when competitive alcohol dehydrogenase antagonists such as ethanol or fomepizole are present.33
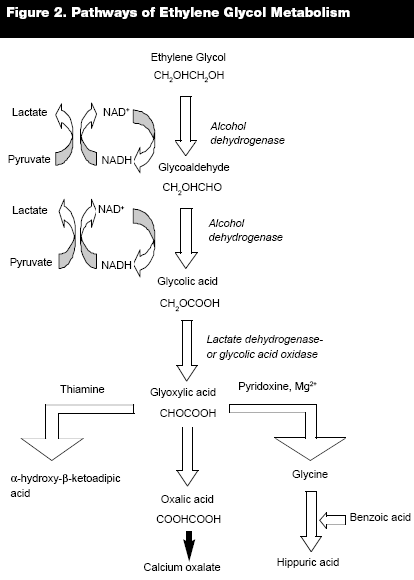
In a manner similar to methanol, ethylene glycol undergoes sequential oxidation to form toxic metabolites. Alcohol dehydrogenase oxidizes the parent compound to glycoaldehyde, which rapidly gets oxidized by aldehyde dehydrogenase resulting in very little appreciable glycoaldehyde being detectable in the blood. The resultant metabolite, glycolic acid, accumulates in substantial amounts due to its relatively slow oxidation to glyoxylic acid and is the primary compound responsible for the metabolic acidosis seen in ethylene glycol poisonings.34 Once formed, however, glyoxylic acid is rapidly oxidized to one of several metabolites, including oxalic acid, which chelates with calcium to precipitate as calcium oxalate crystals.35 (See Figure 2.)
 |
Pathophysiology. The accumulation of glycolic acid correlates with the severity of the metabolic acidosis and depletion of serum bicarbonate concentration seen in ethylene glycol ingestion. Glyoxylic acid and oxalic acid make minimal contributions to the overall derangements in the anion gap as they accumulate at much lower concentrations.36 The successive oxidative metabolic processes result in a depletion of NAD+, leading to inhibition of the citric acid cycle, accumulation of lactic acid, and consequential worsening of the metabolic acidosis.37 The calcium oxalate crystals deposit throughout the body leading to end organ damage. Their accumulation in the epithelium of the proximal renal tubules causes physical blockage and compression of the tubular lumen, which leads to the development of acute renal failure. They also have been found to be directly cytotoxic to the tubules themselves, causing acute tubular necrosis.38 Crystal deposition also has been reported in the brain and may account for multiple cranial nerve abnormalities as well as cerebral edema observed in ethylene glycol poisoned individuals.39 Deposits found in the myocardium on autopsy of ethylene glycol individuals have yet to be linked to any changes in myocardial dysfunction.40 Of note, hypocalcemia resulting from oxalic acid chelation with calcium can lead to prolongation of the QTc interval, negative inotropic effects, dysrhythmias, and seizures; hypocalcemia is likely the cause of acute death in ethylene glycol poisoning.41
Clinical Manifestations. Three classic stages of ethylene glycol poisoning were first described by Kahn and Brotchner in 1950, but the onset, progression, and convergence of these stages varies widely.42 (See Table 2.) Also, the concomitant ingestion of amounts of ethanol may delay the onset of symptoms by several hours, similar to the latency periods seen in methanol/ethanol co-ingestions. The first stage is characterized by neurologic symptoms that occur 30 minutes to 12 hours after ingestion. Ethylene glycol produces transient inebriation and euphoria in a pattern similar to that seen in ethanol intoxication. Slurred speech, ataxia, and nystagmus may occur. However, patients lack the odor of alcoholic beverages and, instead, have a faint, sweet, aromatic odor to them.43 Individuals may also complain of abdominal pain and exhibit nausea and vomiting as ethylene glycol acts directly as a gastric irritant.37 Metabolism produces metabolic acidosis, concurrently, inebriation progresses to lethargy, stupor, and coma. In addition, patients exhibit hyperreflexia, tremors, tetany, and seizures.39 These symptoms typically appear 4-12 hours after ingestion and result from cerebral edema and meningoencephalitis.35,44 Patients may exhibit hyperventilation as a compensatory response to the developing metabolic acidosis.
 |
Cardiopulmonary signs and symptoms are encompassed in the second stage that occurs 12-24 hours post-ingestion. Tachypnea and Kussmall respirations continue in an attempt to correct the severe metabolic acidosis and tachycardia and mild hypertension develop. Hypoxia may become evident, resulting from aspiration, congestive heart failure, or acute respiratory distress syndrome (ARDS); dysrhythmias from hypocalcemia may ensue.45 Death most commonly occurs during this stage, with the etiologies being cardiac dysrhythmias, cerebral edema or infarction, multiple organ failure, or irreversible shock.46
Stage three occurs 24-72 hours after ingestion and is characterized by acute renal failure. Patients who survive the second stage may report severe flank pain and oliguria progressing to anuria.47 Renal dysfunction may be severe enough to require hemodialysis, a therapeutic intervention that usually leads to complete recovery of kidney function. However, some patients sustain permanent damage requiring protracted hemodialysis and, rarely, renal transplantation.37 Cranial neuropathies have been described in the literature, but these presentations usually involve patients who have ingested large quantities of ethylene glycol and present late in their clinical course. These findings typically present themselves 5-20 days after ingestion, undergo slow recovery, but typically resolve after approximately one year.39
Isopropyl Alcohol
History and Epidemiology. Isopropanol, or isopropyl alcohol, is a clear, colorless, volatile alcohol usually sold as a 70% solution in rubbing alcohol, although it is also found in lacquers and used as a solvent in many household, cosmetic, and topical pharmaceutical products. (See Table 1.) Exposure to it is usually through ingestion as an ethanol substitute; however, its toxicity has also been reported via inhalation48 and from dermal absorption in children sponge-bathed with isopropanol to reduce fevers.49,50 In fact, exposures to isopropanol are the most common toxic alcohol exposures reported to poison control centers in the United States.51
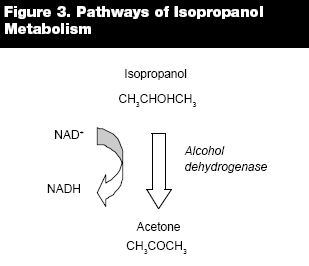
Toxicokinetics and Toxicodynamics. Isopropanol is rapidly absorbed from the stomach after oral ingestion, with peak serum concentrations achieved in 30-60 minutes.52 Metabolism to acetone follows via alcohol dehydrogenase, with ketonemia and ketonuria occurring rapidly after exposure.53 Clearance follows first-order kinetics, and elimination is primarily by the renal route.54 Importantly, metabolism of isopropanol produces not an acid metabolite, but a ketone. (See Figure 3.) In fact, ketosis without acidosis is the defining characteristic of isopropanol poisoning.55
 |
Pathophysiology. Isopropyl alcohol ingestion causes central nervous system (CNS) depression, gastrointestinal irritation, and impaired gluconeogenesis. The hemorrhagic gastritis is thought to result from direct gastric mucosal irritation by the toxic alcohol as opposed to the acetone metabolite.56 However, one author reported a case of hemorrhagic gastritis after dermal exposure, suggesting a possible end-organ effect in contrast to a direct local irritant effect.52 Metabolic acidosis is possible after isopropanol exposure; however, it is the result of isopropanol-induced shock leading to tissue hypoxia and resultant lactate accumulation.48
Clinical Manifestations. Patients suffering from isopropanol intoxication generally present with CNS depression and gastrointestinal distress. The increased lipophilicity of isopropyl alcohol makes it a more potent intoxicant than other alcohols (including ethanol); its effects persist for a longer duration than ethanol due to its slower metabolism as well as the intoxicating effects of acetone.56 Patients also may complain of headache and lightheadedness due to hypotension and may appear ataxic and dysarthric.57 Gastrointestinal symptoms typically include abdominal pain, nausea, vomiting, and hematemesis. The examining physician also may note a characteristic fruity odor to the patient's breath caused by the rapid development of ketonemia.58
Emergency Department Evaluation
Evaluation of individuals who have ingested toxic alcohols should begin with a focused history regarding the time of ingestion, the amount and concentration of the ingested product if known, as well as the nature and onset of symptoms. Information regarding coingestants, particularly ethanol, should also be documented. A review of systems might reveal a feeling of intoxication, visual changes, dyspnea, gastrointestinal symptoms, and flank pain. The physical examination should focus on the vital signs, including respiratory rate, heart rate, and blood pressure. Assessment then should proceed to neurologic status, visual acuity, funduscopic exam, cardiopulmonary status, and abdominal and back tenderness. Initial laboratory data should include complete blood count; serum electrolytes, blood urea nitrogen, creatinine, glucose, liver function tests, lipase and/or amylase, calcium, ketones, and lactate; serum osmolarity; arterial blood gas analysis; and urinalysis. To ensure accurate evaluation of severity of intoxication, ideally serum chemistries, arterial blood gas, and serum osmolality are measured concurrently.
Early in a toxic alcohol overdose, the ingested parent compounds contribute to the measured serum osmolarity, causing it to become elevated. Determination of an osmol gap can thus aid as an early surrogate marker of toxic alcohol poisoning. Serum osmolarity can be calculated using the formula:
2(Na+) + (BUN/2.8) + (glucose/18) + (ethanol/4.6)
The measured osmolarity is typically about 270-290 mOsm/kg H2O. Of note, osmolality or osmolarity should be measured by the freezing point depression method as opposed to the vapor pressure method because the latter underestimates the contribution of volatile alcohols (ethanol, isopropanol, methanol, propylene glycol, and to a lesser extent, ethylene glycol).59 The osmol gap, or the difference between the measure and calculated osmolarity, is then determined using the equation:
Osmol gap (OG) = Omeasured – Ocalculated
A normal osmol gap ranges anywhere between -14 and +10 mOsm; however, individual baseline osmol gaps usually are unavailable for comparison at the time of presentation.60 For example, if an individual's baseline osmol gap is -14 mOsm and on presentation his laboratory results yield an osmol gap of +10 mOsm, his osmol gap on this occasion is 24 mOsm, well beyond the accepted normal range. Therefore, an osmol gap of +10 mOsm cannot exclude the possibility of serious toxic alcohol ingestion. Additionally, a moderately elevated osmol gap (+10 to +20) is not necessarily diagnostic of toxic alcohol poisoning, as other entities such as alcoholic ketoacidosis and lactate-associated acidosis can raise the osmol gap.61 It is the markedly elevated osmol gaps (>25 mOsm) that are difficult to explain by disorders other than toxic alcohol exposures.15
An osmol gap can be used as a rough estimate of the concentration of an alcohol using the following formula
Concentration of alcohol = (osmol gap – 10) x conversion factor
The conversion factor for ethanol is 4.6, methanol 3.2, isopropyl 6.0, and ethylene glycol 6.2. It is important to understand this is a rough estimate for the reasons given above.
As the ethylene glycol and methanol are metabolized, their toxic metabolites, glycolic acid and formic acid, respectively, are formed. This process leads to a decline in serum bicarbonate concentrations and a concomitant rise in serum chloride levels, causing a significant metabolic acidosis before the development of an anion gap. But, as metabolism continues, an anion gap and metabolic acidosis occur together. Glycolic acid and formic acid are the major contributors to the anion gaps in their respective ingestions and the severity of the metabolic acidoses correlate with the serum concentrations of each metabolite.14,62 The size of the anion gap is determined by unmeasured anions. Its value is given by the formula:
Anion Gap (AG) = [(Na+ + K+) – (HCO3- + Cl-)]
A normal anion gap is approximately 12-16 mmol/L, but the actual levels vary between laboratories depending on the accuracy of laboratory measurements.
In addition, an arterial blood gas analysis (ABG) should be performed. Most patients who have a metabolic acidosis from methanol or ethylene glycol poisoning develop a compensatory respiratory alkalosis and an ABG will fully categorize the extent of acidosis and compensatory measures. In general, patients who present with an anion gap of greater than 30 and a serum pH of 7.05 or less must be considered to have ingested a toxic alcohol unless proven otherwise.
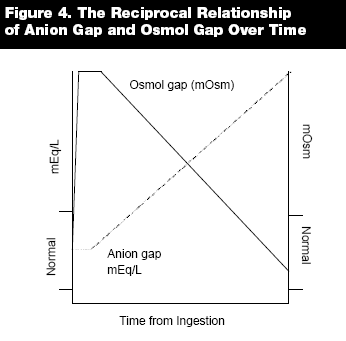
The anion gap and osmol gap have a reciprocal relationship over time. The initial presence of the parent compounds causes an elevated osmol gap, but no contribution to the anion gap. Over time, the formation of acid metabolites contributes to the anion gap and the osmol gap falls as the parent compounds disappear.15 (See Figure 4.)
 |
Another contributor to the anion gap acidosis is the production of lactate. In methanol intoxication, formate inhibits oxidative phosphorylation and leads to anaerobic metabolism. Additionally, the successive oxidative metabolic processes involved in toxic alcohol breakdown result in a depletion of NAD+, leading to inhibition of the citric acid cycle and the favoring of the production of lactic acid from pyruvate.37 Furthermore, hypotension and organ failure in severely poisoned patients also contribute to elevated lactate levels. Interestingly, ethylene glycol poisonings can lead to a false-positive lactate elevation because the glycolic acid metabolite is very similar in structure to lactate and interferes with the measurement of whole blood lactate by some enzymatic methods.27
The determination of serum concentrations of toxic alcohols has been the topic of much debate in the field of toxicology. To begin with, the methodologies typically used are gas chromatography and mass-spectrometry, forms of testing that are expensive and not available in most hospital laboratories. Sending out samples to reference laboratories can result in the withholding of treatment until results are received, leading to possible significant morbidity and mortality. Sample mishandling and improper packaging results in falsely low values due to the high volatility of the compounds.55 Secondly, many patients present late in their exposure and, subsequently, have already metabolized the majority of the parent compounds. It is for this reason that some authors have advocated for the determination of serum glycolic acid and formic acid levels, citing that clinical symptoms and mortality correlate more closely with these serum concentrations and metabolic acidosis rather than with serum ethylene glycol and methanol levels,63,64 but the logistics for obtaining these levels are not currently available in a timely fashion. Lastly, there is significant controversy over the interpretation of serum methanol and ethylene glycol concentrations once they are attained. Historically, 25 mg/dL has been considered a toxic concentration in non-acidotic patients arriving early for care in either methanol or ethylene glycol exposures. However, this is based on older case reports and large outbreaks studies involving bootlegged products, and some textbooks make similar treatment guideline suggestions without reference.9,65 More recent research has demonstrated that in early presenters, a clear acidosis develops only with a methanol level > 126 mg/dL, suggesting that the currently available data are insufficient to apply 25 mg/dL as the treatment threshold. Yet, due to a lack of available prospective studies, this value continues to be used by most experts. Similarly, while there is no specific level of isopropyl alcohol that mandates treatment, some authors recommend that, in a patient that is persistently hypotensive, a level greater than 400 mg/dL requires hemodialysis.66
Urinalysis and microscopy can be helpful in providing evidence of toxic alcohol exposures. The presence of two forms of calcium oxalate crystals, monohydrate and dihydrate, lends supportive proof for ethylene glycol poisoning. The monohydrate, or needle-shaped, form is much more thermodynamically stable than the dihydrate, or envelope-shaped, form. The dihydrate crystals are seen early in ingestions after a latent period of 4-8 hours, but they undergo transformation to the monohydrate crystals within 2-10 hours.14 In the presence of renal failure, these crystals may persist in the urine for 6-10 days.67 The crystal precipitation in the urine leads to the hypocalcemia typically seen in ethylene glycol exposures, and the presence of hypocalcemia and calcium oxalate crystals in the urine is highly suggestive of ethylene glycol poisoning.37 It is to be noted that the amount of oxalate crystalluria does not correlate with the amount of ethylene glycol absorbed and that the presence of these crystals is neither sensitive nor specific to ethylene glycol ingestions.
Sodium fluorescein is added to some commercial antifreeze preparations as a colorant to aid in detection of radiator leaks. Because of this, the use of a Wood's lamp shown on a fresh urine sample can be implemented to aid in the diagnosis of ethylene glycol ingestions. However, there are several points to consider regarding this adjunctive test. First, false-positives may occur if the urine is examined in glass or plastic containers that have inherent fluorescence or if other compounds present in the urine also fluoresce, such as carbamazepine, niacin, and carotene. Secondly, false-negatives may occur if the sample is collected more than four hours after ingestion due to prior excretion of the fluorescein. Finally, extremely acidified urine, such as samples with a pH of less than 4.5, causes the fluorescence of sodium fluorescein to disappear.68 In such cases, the pH needs to be adjusted before ultraviolet light examination. While many authors cite the utility of this test in the determination of ethylene glycol exposures, recent work has refuted it, with one study citing fluorescence of all of their urine specimens.69 It is to be noted that the Wood's lamp also can be used to inspect for spilled sodium fluorescein on the skin and clothing.
Brain imaging can also be a useful component in the evaluation of toxic alcohol exposures. Cranial CT scans may demonstrate evidence of cerebral edema with compression of the supratentorial ventricular system in ethylene glycol toxicity. Cases have also reported reversible hypodensities in the thalamus, basal ganglia, pons, and temporal lobes consistent with meningoencephalitis.70 While CT imaging of the brain often is normal when performed within 24 hours of a methanol exposure, repeat imaging in severe cases most consistently demonstrates bilateral putaminal necrosis with lesser involvement of the caudate nucleus.71 MRI scanning also has been performed in methanol poisonings and has demonstrated atrophy of the optic chiasm as well as persistent occipital lesions.72
Emergency Department Management
Initial treatment strategies for toxic alcohol ingestions should focus on the evaluation and correction of immediate life-threatening complications to the airway, breathing, and circulation. Intravenous access should be established and oxygen applied, and the patient should be placed on hemodynamic monitoring. IV fluid therapy is necessary to ensure adequate urine output, and the volume status of the patient must be monitored closely to prevent fluid overload in those who present with renal dysfunction. Hypoglycemia should be corrected with IV dextrose administration.
Gastrointestinal decontamination methods have never been examined for their efficacy in toxic alcohol ingestions. Many authors cite the rapid absorption of the toxins and the limited binding capacity of activated charcoal to alcohols as reasons for its lack of role. Gastric aspiration and lavage may be effective, and sources support its use if employed one to four hours post-ingestion, especially if moderately large amounts are ingested.73 Activated charcoal also has demonstrated some merit in certain studies and is still recommended at a dosing of 1 g/kg, particularly if a co-ingestant is suspected.74 Syrup of ipecac generally is condemned as a treatment modality due to its sedative properties combined with the risk of aspiration of gastric contents by an obtunded patient as well as its universal ineffectiveness in improving patient outcomes.75
Metabolic acidosis must be treated aggressively as it correlates consistently with severity and outcome, especially in methanol toxicity. Dissociated formic acid, or formate, is much less toxic than undissociated formic acid due to its lower affinity for cytochrome oxidase and its decreased access to the CNS.35 Serum alkalinization also enhances formate clearance in the urine by ion trapping.14 Serum pH values below 7.2 should be managed with intravenous sodium bicarbonate solution to correct the acidosis to above 7.2-7.3. Dosing is calculated using the formula:
NaHCO3 dose = 0.7 x kg x (desired – actual [HCO3-])
When 0.7 is the volume of distribution of bicarbonate in academia, and kg is the lean body weight. Sodium bicarbonate should be mixed with 1/4 NS or D5W up to 2-3 amps per liter.35 Similar dosing can be utilized in metabolic acidosis associated with ethylene glycol poisoning, with the realization that serum alkalinization may worsen hypocalcemia. Of note, bicarbonate can be added to the dialysate bath during hemodialysis to restore the serum bicarbonate concentration.76
The inhibition of alcohol dehydrogenase is at the cornerstone in the management of ethylene glycol and methanol poisoning, and the decision to implement such therapy may be based on the history or on laboratory data alone. Patients with a believable history of ingestion should be treated because, as stated earlier, physical signs and symptoms and laboratory abnormalities initially may be absent. Additionally, patients with an unexplainable osmol or anion gap should also receive treatment. Conversely, isopropyl alcohol exposures should not be treated with ADH inhibitors because acetone is not substantively more toxic than isopropanol.
Historically, ethanol has been used as an antidote for methanol and ethylene glycol poisonings since the 1940s, despite the fact that it has not received FDA approval for such use.77 It is effective because ADH's affinity for ethanol is 15 times greater than its affinity for methanol and 67 times greater than its affinity for ethylene glycol.78,79 Typically, a 10% intravenous solution is administered through a central venous catheter to maintain a serum concentration of 100 mg/dL, although authors have proposed that lower levels could well be as effective based on clinical results.10 Where there is no intravenous alcohol available, oral alcohol can be used, though levels achieved are more variable. The goal is to give 0.6-0.8 g/kg as a loading dose and 0.11-0.15 g/kg/hr as a maintenance dose. This can be accomplished by 0.8-1.0 mL/kg of 95% ethanol in orange juice or by using any common alcoholic beverage using the following conversion:
Grams of ethanol = volume of beverage in mL x 0.9 x proof/200.
Side effects, however, include inebriation and CNS depression, hypoglycemia (more often in children), hyponatremia, thrombophlebitis, pancreatitis, gastritis flushing, and hypotension.44,80 Due to this fact, ethanol has been considered an outdated therapeutic option, and those patients receiving ethanol infusions require intensive care unit admission for close monitoring.
Fomepizole (4-methylpyrazole, 4-MP, Antizol) is an even more potent, recently developed inhibitor of ADH that has received FDA approval for use in methanol and ethylene glycol poisonings. It has an affinity for ADH greater than 8000 times that of ethanol and offers many clinical advantages over ethanol, including a wider therapeutic index, longer duration of action, easier dosing, and more predictable kinetics.81 It reliably inhibits ADH when administered as an intravenous bolus every 12 hours and does not require frequent monitoring and adjustment of plasma concentrations.82,83 Additionally, fomepizole does not carry the side effect profile seen in ethanol administration; the most commonly reported adverse effects are headache, nausea, and dizziness in clinical trials.14,82 The primary disadvantage of fomepizole use is the high acquisition cost (~$1,000 per 1.5-g vial). Assuming you did not admit a patient with ethylene glycol poisoning to the ICU, significant cost savings can be attained through its timely use by the avoidance of ethanol infusion and associated ICU level care, hemodialysis therapy, and total hospital stay. In fact, recent case reports have demonstrated cost reductions of at least 40%.84 The dose of fomepizole is 15 mg/kg intravenously as an initial loading dose followed by 10 mg/kg every 12 hours. After 48 hours, fomepizole induces its own metabolism and the rate must be increased to 15 mg/kg every 12 hours. Of note, if the patient is to require hemodialysis, the dosing frequency should be increased to every 4 hours versus an infusion of 1-1.5 mg/kg/h.37 Patients should continue to receive ethanol or fomepizole therapy until their serum concentration of methanol or ethylene glycol is below 20 mg/dL.82,83
Hemodialysis is the definitive treatment for patients poisoned with toxic alcohols as it clears the parent compounds and their toxic metabolites and corrects the acid-base disorder. Since the advent of fomepizole treatment, however, the indications for hemodialysis have become less clearly defined. Past treatment guidelines recommended hemodialysis for methanol and ethylene glycol levels greater than 50 mg/dL, regardless of patient presentation.81,85 Yet, this concentration alone does not denote toxicity, predict prolonged elimination, or correlate with patient outcome. Patients who present after significant ingestions, but have yet to develop a metabolic acidosis, should have little potential for toxicity if metabolism of their ingestant is effectively blocked with either fomepizole or ethanol.81 In fact, recent evidence suggests that fomepizole monotherapy can be used to safely manage these patients regardless of their initial toxic alcohol concentrations as long as they do not exhibit renal impairment.11,64,86
More recent recommendations for hemodialysis focus on patients who present with end-organ toxicity or severe acidosis, results of toxic metabolites, and problems not addressed by ADH inhibition. Some authors have advocated for attaining serum concentrations of toxic metabolites, attesting that elevated concentrations of formate and glycolic acid better predict ocular injury and renal failure, respectively.63,64 Additionally, patients with renal failure are unable to excrete parent compounds once ADH is blocked, except in expired air in the case of methanol. Hemodialysis, therefore, is suggested in the following conditions: deteriorating vital signs despite intensive supportive care, significant metabolic acidosis (< 7.25–7.30), end-organ toxicity (including vision changes, coma, and seizures), renal failure or electrolyte imbalances unresponsive to conventional therapy.12,37 Extremely high levels of parent compounds or high osmol gaps are considered relative indications for hemodialysis, and decisions must be based on physician judgment and resource availability (such as lack of access to fomepizole). (See Table 3.) Once initiated, therapy should continue until measured parent compound levels are below 20 mg/dL and acidemia is corrected.55 If alcohol concentrations are unavailable, dialysis should continue at least 8 hours or until the osmolal gap is normal (subtracting any possible contribution from ethanol therapy).35 Lastly, as stated earlier, dosing of fomepizole as well as ethanol must be increased during hemodialysis as both compounds are cleared during treatment.
 |
In addition to standard treatment strategies, there are several therapeutic adjuncts that can be employed in toxic alcohol ingestions. Pyridoxine, thiamine, and magnesium are cofactors for the metabolism of ethylene glycol. (See Figure 2.) Pyridoxine promotes its metabolism to glycine, while thiamine and magnesium promote its metabolism to ketoadipate. Similarly, folate and folinic acid, or leukovorin, enhance formic acid metabolism. Case reports have suggested that folate supplementation increases formate clearance in methanol toxicity even more so than hemodialysis.13 Despite these claims, however, no human clinical trials have confirmed the benefit of using these modalities for these purposes.
With regard to hypocalcemia related to ethylene glycol toxicity, calcium should only be replenished if the patient is symptomatic (i.e., cardiac dysrhythmias, tetany, and seizures), as its administration could theoretically enhance calcium oxalate tissue precipitation, worsening toxicity. Lastly, steroids such as methylprednisolone have shown some promise in reducing optic nerve edema and, subsequently, improving visual outcome in methanol toxicity.19,22 Further study needs to be done, however, before widespread use is recommended.
Prognosis and Disposition
In ethylene glycol poisoning, the degree of metabolic acidosis or glycolic acid accumulation at the initiation of treatment closely correlates with the degree of renal injury. One retrospective study found that predictors of renal failure included an initial glycolic acid level of greater than 8 mmol/L, anion gap of greater than 20 mmol/L, or arterial pH of less than 7.30.64 Similarly, a prospective study of methanol ingestions found that endogenous formate concentrations greater than 20 mg/dL were expected to produce ocular injury and acidosis.63 More interestingly, however, were the results of a retrospective review of methanol poisonings, which concluded that coma or seizure on presentation and severe metabolic acidosis, in particular an initial arterial pH < 7, were poor prognostic indicators and carried with them an 84-86% risk of mortality.11
A significant number of ethylene glycol and methanol poisonings require admission to an intensive care unit. Early consultation with a toxicologist or poison control center is strongly encouraged to aid in the diagnosis and care of these individuals, as well as in the coordination of therapeutic options. Poisoned patients often require hemodialysis, a modality that may not be available at some medical centers. Therefore, clinical suspicion must remain high for these clinical entities, and physicians should not hesitate to transfer patients to higher levels of care if necessary.
References
1. Yayci N, Agritmis H, Turla A, et al. Fatalities due to methyl alcohol intoxication in Turkey: An 8-year study. Forensic Sci Int 2003;131: 36-41.
2. Davis LE, Hudson D, Benson BE, et al. Methanol poisoning exposures in the United States: 1993-1998. Clin Toxicol 2002;40: 499-505.
3. IPCS, Health and Safety Guide No. 105. Methanol; World Health Organization: Geneva, 1997;105.
4. Darwish A, Roth CE, Duclos P, et al. Investigation into a cluster of infant deaths following immunization: Evidence for methanol intoxication. Vaccine 2002;20:3585-3589.
5. Frenia ML, Schauben JL. Methanol inhalation toxicity. Ann Emerg Med 1993;22:1919-1923.
6. Swartz R, Millman RP, Billi JE, et al. Epidemic methanol poisoning: Clinical and biochemical analysis of a recent episode. Medicine 1981;60:373-382.
7. Levy P, Hexdall A, Gordon P, et al. Methanol contamination of Romanian home-distilled alcohol. J Toxicol Clin Toxicol 2003;41: 23-28.
8. Graw M, Haffner HT, Althaus L, et al. Invasion and distribution of methanol. Arch Toxicol 2000;74:313-321.
9. Kostic MA, Dart RC. Rethinking the toxic methanol level. J Toxicol Clin Toxicol 2003;41:793-800.
10. Palatnick W, Redman LW, Sitar DS, et al. Methanol half-life during ethanol administration: Implications for management of methanol poisoning. Ann Emerg Med 1995;26:202-207.
11. Liu JL, Daya MR, Carrasquillo O, et al. Prognostic factors in patients with methanol poisoning. J Toxicol Clin Toxicol 1998;36: 175-182.
12. Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. Clin Toxicol 2002;40:415-446.
13. Kerns II W, Tomaszewski C, McMartin K, et al. Formate kinetics in methanol poisoning. Clin Toxicol 2002;40:137-143.
14. Jacobsen D, Webb R, Collins TD. Methanol and formate kinetics in late diagnosed methanol intoxication. Med Toxicol 1988;3:418-423.
15. Hovda KE, Hunderi OH, Rudberg N, et al. Anion and osmolal gaps in the diagnosis of methanol poisoning: Clinical study in 28 patients. Intensive Care Med 2004;30:1842-1846.
16. Jacobsen D, Jansen H, Wiik-Larsen E, et al. Studies on methanol poisoning. Acta Med Scand 1982;212:5-10.
17. Scholl P. Formate as an inhibitor of cytochrome c oxidase. Biochem Biophys Res Com 1975;67:610-616.
18. Treichel JL, Henry MM, Skumatz CMB, et al. Formate, the toxic metabolite of methanol, in cultured ocular cells. Neurotoxicol 2003; 24:825-834.
19. Halavaara J, Valanne L, Setala K. Neuroimaging supports the clinical diagnosis of methanol poisoning. Neurorad 2002;44:924-928.
20. Ariwodola OJ, Weiner JL. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: Role of presynaptic GABA(B) receptors. J Neurosci 2004;24:10679-10686.
21. Grobin AC, Matthews DB, Devaud LL, et al. The role of GABAA receptors in the acute and chronic effects of ethanol. Psychopharm 1998;139:2-19.
22. Onder F, Ilker S, Kansu T, et al. Acute blindness and putaminal necrosis in methanol intoxication. Int Ophthal 1999;22:81-84.
23. Hantson P, Mahieu P. Pancreatic injury following acute methanol poisoning. Clin Toxicol 2000;38:297-303.
24. Grufferman S, Morris D, Alvarez J. Methanol poisoning complicated by myoglobinuric renal failure. Am J Emerg Med 1985;3:24–26.
25. Ley CO, Gali FG. Parkinsonian syndrome after methanol intoxication. Eur Neurol 1983;22:405–409.
26. Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 annual report of the American Association of Poison Control Centers' national poisoning and exposure database. Clin Toxicol 2006;44:803-932.
27. Woo MY, Greenway DC, Nadler SP, et al. Artifactual elevation of lactate in ethylene glycol poisoning. J Emerg Med 2003;25: 289-293.
28. Litovitz TL, Klein-Schwartz W, Dyer KS, et al. 1997 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 1998;16:443-497.
29. Driver J, Tardiff RG, Sedik L. In vitro percutaneous absorption of [14C] ethylene glycol by man. J Exp Anal Env Epid 1993;3: 277-284.
30. Gessner PK, Parke DV, Williams RT. Studies in detoxification. 86. The metabolism of 14C-labelled ethylene glycol. Biochem J 1961; 79:482-489.
31. Cheng JT, Beysolow TD, Kaul B, et al. Clearance of ethylene glycol by kidneys and hemodialysis. J Toxicol Clin Toxicol 1987;25: 94-108.
32. Eder AF, McGrath CM, Dowdy YG, et al. Ethylene glycol poisoning. Toxicokinetic and analytical factors affecting laboratory diagnosis. Clin Chem 1998;44:168–177.
33. Peterson DC, Collins, AJ, Himes JM, et al. Ethylene glycol poisoning. Pharmacokinetics during therapy with ethanol and hemodialysis. N Engl J Med 1981;304:21–23.
34. Jacobsen D, Hewlett TP, Webb R, et al. Ethylene glycol intoxication: Evaluation of kinetics and crystalluria. Am J Med 1988;84: 145-152.
35. Jacobsen D, McMartin KE. Methanol and ethylene glycol poisonings: Mechanisms of toxicity, clinical course, diagnosis and treatment. Med Toxicol 1986;1:309-334.
36. Moreau CL, Kerns II W, Tomaszewski CA. Glycolate kinetics and hemodialysis clearance in ethylene glycol poisoning. J Toxicol Clin Toxicol 1998;36:659-666.
37. Barceloux DG, Krenzelok EP, Olson K, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. Clin Toxicol 1999;37:537-560.
38. Guo C, Cenac TA, Li Y, et al. Calcium oxalate, and not other metabolites, is responsible for the renal toxicity of ethylene glycol. Toxicol Let 2007;173:8-16.
39. Spillane L, Roberts JR, Meyer AE. Multiple cranial nerve deficits after ethylene glycol poisoning. Ann Emerg Med 1991;20:208-210.
40. Introna F Jr, Smialek JE. Antifreeze (ethylene glycol) intoxications in Baltimore. Report of six cases. Acta Morphol Hung 1989;37: 245-263.
41. Scully R, Galdabini J, McNealy B. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 38-1979. N Engl J Med 1979;301:650-657.
42. Kahn HS, Brotchner RJ. A recovery from ethylene glycol (antifreeze) intoxication; a case of survival and two fatalities from ethylene glycol including autopsy findings. Ann Intern Med 1950;32:284–294.
43. Burns MJ. Ethylene Glycol. In: Wolfson AB, et al, eds. Harwood-Nuss' Clinical Practice of Emergency Medicine. Philadelphia: Lippincott Williams and Wilkins Co.;2005:1454-1458.
44. Jacobsen D, McMartin KE. Antidotes for methanol and ethylene glycol poisoning. J Toxicol Clin Toxicol 1997;35:127-143.
45. Catchings TT, Beamer WC, Lundy L, et al. Adult respiratory distress syndrome secondary to ethylene glycol ingestion. Ann Emerg Med 1985;14:594–596.
46. Gordon HL, Hunter JM. Ethylene glycol poisoning. Anaesthesia 1982;37:332–338.
47. Bobbit WH, Williams RM, Freed CR. Severe ethylene glycol intoxication with multisystemic failure. West J Med 1986;144:225–228.
48. Vicas IMO, Beck R. Fatal inhalational isopropyl alcohol poisoning in a neonate. J Toxicol Clin Toxicol 1993;31:473-481.
49. Lewin GA, Oppenheimer PR, Wingert WA. Coma from alcohol sponging. JACEP 1977;6:165-167.
50. Vivier PM, Lewander WJ, Martin HF, et al. Isopropyl alcohol intoxication in a neonate through chronic dermal exposure: A complication of a culturally based umbilical care practice. Ped Emerg Care 1994;10:91-93.
51. Watson WA, Litovitz TL, Klein-Schwartz W, et al. 2003 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2004;22:335-404.
52. Dyer S, Mycyk MB, Ahrens WR, et al. Hemorrhagic gastritis from topical isopropanol exposure. Ann Pharmacother 2002;36:1 733-1735.
53. Lacouture PG, Heldreth DD, Shannon M, et al. The generation of acetomemia/acetonuria following ingestion of a subtoxic dose of isopropyl alcohol. Am J Emerg Med 1989;7:38-40.
54. Pappas AA, Ackerman BH, Olsen KM, et al. Isopropanol ingestion: A report of 6 episodes with isopropanol and acetone serum concentration time date. J Toxicol Clin Toxicol 1991;29:11-21.
55. Weiner SW. Toxic Alcohols. In: Flomenbaum NE, et al, eds. Goldfrank's Toxicologic Emergencies. New York: McGraw-Hill Co.;2002:1447-1459.
56. Ford MD. Isopropanol. In: Ford MD, et al, eds. Clinical Toxicology. Philadelphia: WB Saunders Co.;2001:769-773.
57. Rosansky SJ. Isopropyl alcohol poisoning treated with hemodialysis: kinetics of isopropyl alcohol and acetone removal. J Toxicol Clin Toxicol 1982;19:265-271.
58. Suchard JR. Isopropanol, acetone, and other glycols. In: Wolfson AB, et al, eds. Harwood-Nuss' Clinical Practice of Emergency Medicine. Philadelphia: Lippincott Williams and Wilkins Co.;2005:1461-1464.
59. Eisen TF, Lacouture PG, Woolf A. Serum osmolality in alcohol ingestions: Differences in availability among laboratories of teaching hospital, non-teaching hospital, and commercial facilities. Am J Emerg Med 1989;7:256–259.
60. Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: Normal values and limitations. J Toxicol Clin Toxicol 1993; 31:81-94.
61. Schelling JR, Howland RL, Winter SD, et al. Increased osmolal gap in alcoholic ketoacidosis and lactic acidosis. Ann Intern Med 1990; 113:580-582.
62. Hewlett TP, McMartin KE, Lauro AJ, et al. Ethylene glycol poisoning: The value of glycolic acid determinations for diagnosis and treatment. J Toxicol Clin Toxicol 1986;24:389–402.
63. Osterloh JD, Pond SM, Grady S, et al. Serum formate concentrations in methanol intoxication as a criterion for hemodialysis. Ann Intern Med 1986;104:200-203.
64. Porter WH, Rutter PW, Bush BA, et al. Ethylene glycol toxicity: The role of serum glycolic acid in hemodialysis. Clin Toxicol 2001; 39:607-615.
65. Bennett IL, Cary FH, Mitchell GL Jr, et al. Acute methyl alcohol poisoning: A review based on experiences in an outbreak of 323 cases. Medicine (Baltimore) 1953;32:432-463.
66. Church AS, Witting MD. Laboratory testing in ethanol, methanol, ethylene glycol, and isopropanol toxicities. Clin Lab Emerg Med 1997;15:687-692.
67. Parry MF, Wallach R. Ethylene glycol poisoning. Am J Med 1974; 57:143–150.
68. Winter ML, Ellis MD, Snodgrass WR. Urine fluorescence using a Wood's lamp to detect the antifreeze additive sodium fluorescein: A qualitative adjunctive test in suspected ethylene glycol ingestions. Ann Emerg Med 1990;19:663-667.
69. Casavant MJ, Shah MN, Battels R. Does fluorescent urine indicate antifreeze ingestion by children? Pediatrics 2001;107:113-114.
70. Maier W. Cerebral computed tomography of ethylene glycol intoxication. Neuroradiology 1983;24:175–177.
71. Patankar T, Bichile L, Karnad D, et al. Methanol poisoning: Brain computed tomography scan findings in four patients. Aust Rad 1999;43:526-528.
72. Hsu HH, Chen CY, Chen FH, et al. Optic atrophy and cerebral infarcts caused by methanol intoxication: MRI. Neurorad 1997; 39:192–194.
73. Elwell RJ, Darouian P, Bailie GR, et al. Delayed absorption and postdialysis rebound in a case of acute methanol poisoning. Am J Emerg Med 2004;22:126-127.
74. Decker WJ, Corby DG, Hilburn RE, et al. Adsorption of solvents by activated charcoal, polymers, and mineral spirits. Vet Hum Toxicol 1981;23:44–46.
75. Krenzelok EP, McGuigan M, Lheur P. Position statement: Ipecac syrup. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. J Toxicol Clin Toxicol 1997;35:699–709.
76. Ward KW, Perkins RA, Kawagoe JL, et al. Comparative toxicokinetics of methanol in the female mouse and rat. Fundam Appl Toxicol 1995;26:258–264.
77. Roe O. Methanol poisoning: Its clinical course, pathogenesis, and treatment. Acta Med Scand 1946;126 (Suppl 182):1-253.
78. Pietruszko R. Human liver alcohol dehydrogenase inhibition of methanol activity by pyrazole, 4-methylpyrazole, 4-hydroxymethylpyrazole and 4-carbopyrazole. Biochem Pharmacol 1975; 24:1603-1607.
79. Pietruszko R, Voigtlander K, Lester D. Alcohol dehydrogenase from human and horse liver-substance specificity with diols. Biochem Pharmacol 1978;27:1296-1297.
80. Roy M, Bailey B, Chalut D, et al. What are the adverse effects of ethanol used as an antidote in the treatment of suspected methanol poisoning in children? J Toxicol Clin Toxicol 2003;41:155-161.
81. Sivilotti MLA, Burns MJ, McMartin KE, et al. Toxicokinetics of ethylene glycol during fomepizole therapy: Implications for management. Ann Emerg Med 2000;36:114-125.
82. Brent J, McMartin J, Phillips S, et al. Fomepizole for the treatment of ethylene glycol poisoning. N Engl J Med 1999;340:832-838.
83. Brent J, McMartin K, Phillips S, et al. Fomepizole for the treatment of methanol poisoning. N Engl J Med 2001;344:424-429.
84. Boyer EW, Mejia M, Woolf A, et al. Severe ethylene glycol ingestion treated without hemodialysis. Pediatrics 2001;107:172-173.
85. Gonda A, Gault H, Churchill D, et al. Hemodialysis for methanol intoxication. Am J Med 1978;64:749-758.
86. Hantson P, Hassoun A, Mahieu P. Ethylene glycol poisoning treated by IV 4-methylpyrazole. Intensive Care Med 1998;24:736–739.
Exposures to toxic alcohols such as methanol, ethylene glycol, and isopropanol have been reported in the medical literature for decades. These agents are found in a variety of household products, leading to accidental ingestion in the pediatric population and intentional ingestion in the adult population as a suicidal agent or as an inexpensive substitute for ethanol.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.