Sickle Cell Disease in the Emergency Department
AUTHORS
Erica Bates, MD, Assistant Professor, Department of Emergency Medicine, Penn State Health, Hershey Medical Center, Hershey, PA
Matthew Turner, MD, Department of Emergency Medicine, Penn State University
PEER REVIEWER
Catherine A. Marco, MD, FACEP, Professor, Department of Emergency Medicine, Penn State Health, Hershey Medical Center, Hershey, PA
Introduction
Sickle cell disease (SCD) is a multi-system pathology that presents with episodes of acute illness and progressive organ damage.1 Far from being a single disease, SCD is associated with multiple genotypes that share a common tendency for erythrocytes to deform into crescent or sickle shapes.2 In the most common form, the patient inherits a mutated copy of the β-globin gene from both parents, causing a homozygosity of an abnormal form of hemoglobin called HbS.3 Ultimately, this leads to a malformation in the patient’s hemoglobin molecules in their red blood cells (RBCs), which leads to erythrocyte malformation into the classic sickle shape.4 This malformation can lead to a plethora of downstream effects, which can affect nearly every organ system in the body, on average leading to a life expectancy 20 years shorter than the general population.1
Patients who have one HbS gene and one normal copy of the gene usually are asymptomatic carriers and are unlikely to experience clinical manifestations in most normal conditions due to the amount of normal hemoglobin still produced by the unaffected copy of the gene. These heterozygous patients are not usually included when discussing patients who are at risk of serious complications from SCD. Individuals who inherit one HbS gene and one hemoglobin C gene, another type of mutated β-globin allele, often experience a milder form of symptomatic SCD. These patients may experience the same complications as patients with two copies of the HbS gene, such as acute pain crises, but often have less severe or less frequent symptoms.1
Sickle cell anemia is a highly variable disease (see Table 1), with wide ranges of pathology and presentation. High levels of fetal hemoglobin and factors such as the α-thalassemia trait have been found to be protective factors. Up to 39% of patients may have no pain episodes within the past year, while 1% will have had more than six episodes of pain within the past year.4 Fortunately, SCD can be diagnosed easily through inexpensive and widely available hemoglobin electrophoresis. Many countries, including the United States and the United Kingdom, have enacted universal neonatal screenings for the disease, with a significant improvement in pediatric mortality.4
Table 1. Acute Sickle Cell Disease Problems in the Emergency Department |
|||
Problem |
Typical Presentation |
Diagnosis |
Treatment |
Acute pain crisis |
|
|
|
Avascular necrosis |
|
|
|
Infection |
|
|
|
Acute chest syndrome |
|
|
|
Stroke |
Focal neurological deficits |
|
|
Acute anemia |
Decrease in hemoglobin |
|
|
Priapism |
|
|
|
SCD is a growing concern in global health. More than 300,000 infants are born with this condition every year, with particularly high levels in sub-Saharan Africa, the Middle East, India, and the Mediterranean region. This geographic distribution is due in large part to the significant protection that SCD heterozygosity offers against endemic malaria. However, malaria in a patient with SCD homozygosity causes severe anemia and higher mortality rates. The prevalence of SCD outside of these regions is increasing as well; more than 100,000 patients in the United States have some form of SCD.1
Despite the growing number of individuals living with SCD, the number of U.S. hematologists specifically trained in the management of SCD is decreasing.5 In the emergency department (ED) setting, significant gaps in care exist, making it imperative that emergency physicians know how to manage this complex disease process. SCD often is stigmatized; patients with SCD frequently are perceived as drug-seeking, and therefore pain associated with complications of the disease often is undertreated.6 Patients with SCD often perceive the healthcare system with mistrust, seeing it as a place of stigma and bias against them. This is further exacerbated by the historical inequalities and structural racism that many Black patients have experienced in the U.S. healthcare system.1 Health disparities are significant for SCD patients, who have a notable gap in life expectancy compared to the average American.7 U.S. EDs receive more than 200,000 visits tied to SCD per year, and emergency physicians must be skilled in the treatment of this unique and vulnerable patient population to avoid exacerbating the high rates of morbidity and mortality.8
Acute Pain Crisis
Sickle cell pain crises are caused by a complex interaction of deoxygenated HbS leading to vaso-occlusion in the patient’s microcirculation. Sickle-shaped erythrocytes adhere to the endothelium and lead to microvascular occlusion. The resulting hypoxia, tissue ischemia, and an inflammatory cascade lead to a feedback loop of abnormal blood flow and worsening pain.9 Reperfusion subsequently can exacerbate this process.10 The diagnosis of sickle cell pain crisis is clinical and relies completely on the patient’s report of acutely increased pain; laboratory values and other objective findings should be obtained only to rule out other complications, such as acute chest syndrome.1 Laboratory values may be unchanged from the patient’s baseline during an acute pain crisis.
Up to 67% of patients with SCD will delay presenting to the ED due to a combination of factors, including a history of stigma and perceptions of mistreatment.8 However, one study found that when SCD patients do visit the ED, 78% present with the chief complaint of acute pain crisis. Sickle cell pain crises are the most common cause of hospital admissions for this patient population.11 Patients may describe a “pounding, cutting, gnawing, or like a generalized toothache” pain with a prodrome of one to two days. This pain typically will peak on day 3, and will last to day 6 or 7 before resolving. A hospital stay of nine to 11 days is average for this patient population.12 Pain crises may be triggered by a wide range of etiologies, including fever, hypoxia, acidosis, dehydration, pregnancy, menstruation, obstructive sleep apnea, alcohol consumption, diabetes, and even cold weather, with higher wind speeds corresponding to an increased rate of hospital admissions for SCD patients.3,12
Pain crises are associated with an increased risk of death and a significantly increased risk of complications such as acute chest syndrome.9,12 On presentation to the ED, these patients should be treated with adequate pain control, which will require individualized dosing and titration. If possible, medications that were effective during past pain crises should be used to determine appropriate dosing. Some patients may present with a personalized pain control care plan developed by their primary hematologist. Treatment should be initiated quickly, within one hour of arrival if possible.1 Opioids, as well as intravenous (IV) hydration, antihistamines as needed for opioid-related itching, antiemetics as needed for nausea, and oxygen to address hypoxia also should be used to achieve symptom control. Individualized dosing of analgesics has been shown to be significantly more effective at reducing hospital admissions than weight-based dosing of analgesics.1 Nonsteroidal anti-inflammatory drugs (NSAIDs) may be helpful, but they should be administered carefully, given the nephrotoxicity of these drugs and the risk of renal impairment in SCD.12 The National Heart, Lung, and Blood Institute (NHLBI) recommends an aggressive regimen of pain control, with patient-controlled early analgesia and reassessments of the patient every 15-30 minutes.13 Preventive treatments, such as hydroxyurea to increase fetal hemoglobin, stem cell transplantation, gene therapy, and antibodies such as crizanlizumab can be useful to prevent or reduce the frequency of sickle cell pain crises, but are unlikely to be helpful in the acute ED setting.2,9
In patients presenting in acute pain crisis, it is important that orthopedic complications of SCD, including bone marrow infarction and avascular necrosis, be ruled out by the emergency physician.12 The patient’s history should take initial priority: If the patient reports that their pain crisis is different from previous pain crises, a full work-up should be done to avoid the risk of cognitive bias and the potential for another acute condition being present.10 (See Figure 1.) Unfortunately, a 2021 multicenter prospective cohort study found that SCD patients in acute pain crises receive substantially better outcomes via treatments in infusion centers than in an ED.14 More work is needed to improve the quality and consistency of emergency care for patients experiencing acute pain crisis.
Figure 1. Clinical Management of Pain and Other Aspects of Sickle Cell Disease |
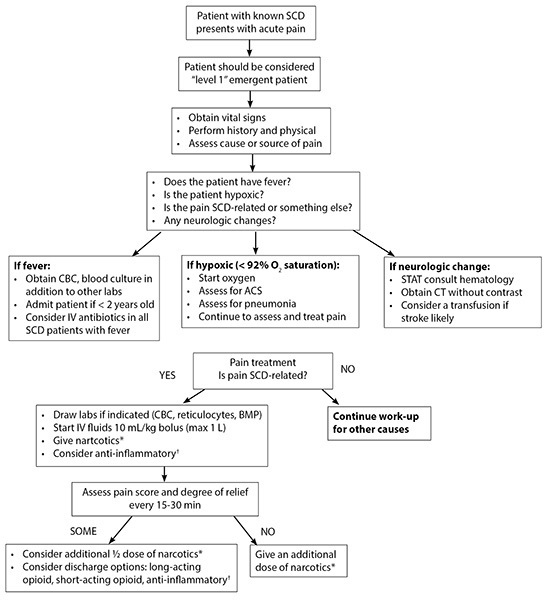 |
* Morphine 0.1 mg/kg or hydromorphone 0.015-0.02 mg/kg. † Ketorolac (Toradol®) 0.5 mg/kg (max of 30 mg) or ibuprofen (10 mg/kg). ACS: acute chest syndrome; BMP: basic metabolic panel; CBC: complete blood count; CT: computed tomography; IV: intravenous; SCD: sickle cell disease; STAT: immediate. Reprinted with permission from: Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Rev 2013;27:279-287. |
Avascular Necrosis
Avascular necrosis occurs in patients with SCD when bone tissue dies as a result of tissue ischemia from compromised microcirculation. Up to 22% of patients with SCD may experience avascular necrosis, usually in the hips, shoulders, and spine.1 This significant disease process can lead to eventual collapse of the bony structures. Joint pain and limited range of motion are the most common symptoms, although some radiologically detected avascular necrosis also may be asymptomatic.15 Treatment includes physical therapy as well as potential orthopedic procedures such as hip coring — surgical removal of dead bone from the femoral head, sometimes in conjunction with injection of bone marrow aspirate.1 Some patients require arthroplasty.15 Teenagers and young adults are at particular risk of avascular necrosis of the femoral head. Young children can even experience cranial bony infarcts, which may be confused with acute stroke due to the presence of neurological symptoms.7
Pain due to avascular necrosis may be confused with an acute pain crisis. It also is important to consider septic arthritis in the differential diagnosis for acute onset monoarticular joint pain in a patient with SCD presenting to the ED. Earlier in the process, avascular necrosis is best seen on T2-weighted magnetic resonance imaging (MRI) images; however, as the disease progresses, advanced changes may be observed on simple radiographs as well.16 In the ED, the initial care of avascular necrosis is largely supportive, with adequate analgesia and referral to appropriate orthopedic and hematology specialist care.
Infection
Patients with SCD are significantly more vulnerable to multiple bacterial infections than the general population, in large part because of the hyposplenism seen in this disease process. Over time, repeated episodes of sickling and ischemic damage, as well as damage to arterioles, leads to functional infarcts of the spleen. This causes the body to become particularly vulnerable to encapsulated pathogens, such as Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis, and salmonellae. Approximately 6% of SCD patients present to the ED citing fever or infection as their chief complaint.11 Emergency physicians should be aware of the unique infectious challenges facing this patient population.
Children with SCD are especially at risk for serious infections, even from non-encapsulated bacteria and viruses, suggesting that there are further defects in the immune system. Even respiratory illnesses, such as respiratory syncytial virus and influenza, may lead to life-threatening complications, such as acute chest syndrome, aplastic crisis, or a bacterial superinfection. Given this increased risk of morbidity and mortality, any fever in a pediatric patient with SCD should be treated as an emergency. Evaluation should include a thorough history and physical examination, chest X-ray to exclude pneumonia or acute chest syndrome, complete blood count, reticulocyte count, blood cultures, empiric broad-spectrum intravenous antibiotics with gram-negative coverage such as ceftriaxone, and consideration of hospitalization for further observation.17
Patients with SCD have an increased predisposition to osteomyelitis due to vaso-occlusion and infarction in the bones and are at risk for aplastic crisis due to viruses such as parvovirus B19.7 Patients should be thoroughly educated on preventive measures, including to protect against Salmonella by particular care when cooking or preparing chicken and eggs.18 Zinc supplementation may be helpful, as well as following recommended vaccination programs.17 In pediatric populations, oral prophylactic penicillin has been found to significantly reduce the risk of pneumococcal infections. Controversy remains about whether this penicillin prophylaxis can be safely discontinued at the age of 5 years, or whether it should be continued lifelong for asplenic patients, with multiple organizations offering differing guidelines.19 Erythromycin may be used as an alternative in cases of penicillin allergy.20
Acute Chest Syndrome
Acute chest syndrome is a leading cause of death in SCD patients.7 A complex pathology with a number of causes, including infection, pulmonary infarction, infection, and fat embolism, acute chest syndrome strongly resembles bacterial pneumonia, with fever, elevated white blood cells (WBCs), pleuritic chest pain, pleural effusions, and cough.21 Approximately half of all SCD patients will experience at least one episode of acute chest syndrome during their lifetimes. Ten percent to 20% of hospitalized patients with SCD develop acute chest syndrome.12 Often patients will develop acute chest syndrome around day 2.5 of hospitalization for an acute pain crisis, although it can occur at any time.22
Acute chest syndrome typically presents with nonspecific symptoms ranging from fever, cough, and chest pain to shortness of breath, hemoptysis, and fever. Clinically, a physical exam is unreliable; one study found more than 60% of acute chest syndrome cases were not clinically suspected until the diagnosis was confirmed by routine radiographs.23 Therefore, it is important that chest X-rays be included in evaluating SCD patients who present to the ED with fever, chest pain, or respiratory symptoms. If patients display fever; hypoxemia; tachypnea; or cough, chest pain, or wheezing, in addition to new radiodensities on chest X-ray, they should be diagnosed with acute chest syndrome.21
Treatment for acute chest syndrome is largely supportive. Patients should be given supplemental oxygen as needed for hypoxemia, pain control, empiric broad-spectrum antibiotics, hydration via IV fluids (typically D5 in 0.45% normal saline at 1.5 times maintenance fluid rate), incentive spirometry, diuretics if pulmonary edema is present, and blood transfusions as needed.21 Transfusions may be accomplished through simple transfusion or exchange transfusion. Early consultation with a hematologist should be obtained to determine the best transfusion strategy. Exchange transfusion sometimes is required for rapid dilution of HbS and requires the use of dedicated nursing staff and specialized resources.22 NSAIDs should be avoided, since they exacerbate acute chest syndrome symptoms.23 In severe cases, patients may require mechanical ventilation. Ultimately, patients with presentations concerning for acute chest syndrome should be treated empirically and admitted to an appropriate level of care where hematology consultation is available, given the high mortality of this pathology.
Stroke
Twenty-four percent of patients with SCD experience a stroke by age 45 years.24 The greatest risk of stroke occurs between the ages of 2 and 5 years, with a 1% incidence per year in these patient populations. These events often manifest with neurocognitive difficulties and risk of further brain infarcts.4,17 Intracranial hemorrhages occur most commonly in patients with SCD between 20 and 30 years of age. Patients who have experienced one stroke have a 60% chance of recurrence.4
Preventive measures should be highly emphasized in this patient population, with transcranial Doppler studies of the middle cerebral arteries used to identify high-risk pediatric patients.17 Chronic transfusions to lower HbS below 30% are a possible means of treatment, but they carry the risk of iron overload and alloimmunization.25 There also appears to be a dangerously high risk of stroke recurrence after chronic transfusion is discontinued.24 A chronic transfusion regimen also often requires a central venous catheter, which leads to an increased risk of infections and thrombosis.1 Recent data from the Stroke Prevention in Nigeria (SPRING) trial suggest that low- and moderate-dose hydroxyurea can be safely used to prevent stroke in SCD.25
In the ED, patients with SCD should be evaluated for stroke based on their clinical presentation. Computed tomography (CT) imaging may be used to identify an acute hemorrhagic stroke, which is more likely in adults, but is unlikely to detect the acute ischemic strokes, which are more likely in pediatric patients. When ischemic stroke is suspected after a negative head CT, urgent MRI and/or MR angiography of the brain also should be obtained, if possible.26
Exchange transfusion is the primary form of treatment in pediatric ischemic strokes.1 Adult patients with SCD presenting with ischemic stroke should receive standard stroke management, with the use of thrombolytics if indicated, in addition to exchange transfusion.1 Emergency physicians should consult with neurology and hematology specialists as early as possible, given the complex care that these patients require. Patients should be transferred to dedicated stroke units if possible, given the importance of supportive measures to optimize their care for the first 24 hours.26
Data for treatment of hemorrhagic stroke in SCD are scant. The value of exchange transfusions in these scenarios remains unclear.1 These patients should be transferred to an intensive care unit with reversal of any anticoagulation agents, administration of antiepileptics, management of blood pressure, and possible neurosurgical intervention in keeping with the standard of care for all hemorrhagic stroke patients.26 Hematology and neurosurgery should be consulted early to closely follow these patients.
In all cases of stroke, it is recommended that adults and children with SCD be transferred to a dedicated stroke unit with neurology and hematology services available, with a recovery plan for long-term rehabilitation and follow-up. This is especially important in pediatric populations, given the significant risk of recurrence and the high degree of cognitive impairment that may occur due to these strokes.26
Acute Anemia
Hemolytic anemia is one of the major pathophysiological processes associated with SCD. Acute anemia is defined as an acute decrease in the patient’s hemoglobin from their baseline by 2 g/dL or greater.1 HbS polymerization can lead to increased levels of hemolysis, which in the short term can lead to acute anemia. Chronically, this also may lead to the release of free radicals, vasculopathy, systemic and pulmonary hypertension, and diffuse dysfunction in the walls of blood vessels.4 In the ED setting, acute anemia should be considered in patients with sickle cell anemia who present with dyspnea, chest pain, presyncope/lightheadedness, or fatigue.
The most common cause of acute anemia in pediatric patients is splenic sequestration, which has a lifetime prevalence of up to 30% and may occur in children as young as 2 months old.1 Patients experiencing acute splenic sequestration often will present with left upper quadrant abdominal pain, thrombocytopenia, anemia, and a 25% increase in reticulocyte count as the body attempts to compensate for the sequestered RBCs.1 These patients should be treated with emergency transfusions and then closely monitored for hyperviscosity syndrome as trapped blood is released from the spleen. In the most severe cases, this may lead to ischemic damage of multiple organs, including the brain.1 Splenectomy may be indicated in these cases due to the high risk of recurrence.1
Acute anemia also may be caused by aplastic crises, which can be triggered by infections such as parvovirus B19.1 As the name suggests, an aplastic crisis occurs when the bone marrow stops producing new red blood cells. An inappropriately low reticulocyte count suggests the presence of an aplastic crisis. In SCD patients, this is particularly dangerous because sickle-shaped erythrocytes survive only 10-14 days (as opposed to 120 days for normal cells) and must be replaced continuously to maintain a stable hemoglobin.1 Transfusion is the mainstay of treatment; these patients should be monitored closely in the subsequent weeks due to the possibility of a delayed hemolytic transfusion reaction.1 Iron chelation also should be considered, due to the possibility of iron overload from a chronic transfusion regimen.22 Hemoglobin concentrations generally should be corrected only to the patient’s baseline value, unless otherwise recommended by the patient’s hematologist.2
Pregnancy
Pregnancy in SCD is associated with a wide range of complications, including increased pain, susceptibility to infections, acute chest syndrome, and increased risk of thrombosis.1 Vaso-occlusive crises may lead to acute chest syndrome, necrosis, and impaired blood flow to the fetus, leading to fetal hypoxia and poor fetal outcomes.27 Pregnant women also have a higher risk of preeclampsia and eclampsia, as well as higher incidences of severe anemia, severe infections, pulmonary embolism, and preterm birth.1 Postpartum women in particular have an increased risk of mortality following complications from SCD.2
Unfortunately, hydroxyurea is a potentially teratogenic medication, and, therefore, it is recommended to discontinue the drug at least three months before conception.27 Care during pregnancy should be supportive and involve close follow-up. Delivery should be conducted in centers equipped to manage high-risk pregnancies, with the recommended use of epidural anesthesia and continuous fetal heart rate monitoring due to the elevated risk of fetal distress. After delivery, low molecular weight heparin is recommended for seven days following vaginal delivery and six weeks following a cesarean delivery, due to the increased risk of venous thromboembolism (VTE) in postpartum women with SCD.27
Venous Thromboembolism
SCD is associated with a hypercoagulable state, endothelial dysfunction, and increased blood stasis. This leads to a higher risk of VTE, including deep venous thrombosis and pulmonary embolus (PE). Pregnant and postpartum women are at particular risk from these complications.28 Pregnant and postpartum women who present with chest pain and respiratory distress with negative chest X-rays should be evaluated for PE. In these cases, low molecular weight heparin should be initiated even before the diagnosis is confirmed.27
Priapism
Forty percent of male patients with SCD will experience priapism, an erection lasting longer than four hours.1 Priapism due to SCD is caused by a low-flow ischemic state and may occur in both pediatric and adult patients. It may occur as a single episode or as “stuttering priapism,” which is caused by repetitive vaso-occlusion, and presents as recurrent episodes that last from several minutes to several hours.29 RBC sickling causes blood to sludge in the corpora cavernosa. Nocturnal occurrences are common, possibly due to a combination of nighttime hypoventilation leading to a mild acidosis, which induces erythrocyte sickling, and due to nocturnal tumescence compressing the venous channels of the corpora cavernosa.29 Priapism is a urologic emergency: it has a five-fold increased risk of severe erectile dysfunction over the general male population and is closely associated with significant mental health sequelae, including anxiety and depression. In severe untreated cases, penile compartment syndrome may result.30
Painful episodes of penile erection that have lasted less than three hours may be empirically treated in the ED first. Acute management may be as simple as cold/warm compresses, hot showers, analgesia, and rehydration. If the priapism fails to resolve, urology should be consulted urgently. Corporal aspiration may be required; in the most severe cases, shunt surgery may be needed.30 In cases of priapism that have lasted longer than three hours prior to presentation, urology should be consulted immediately. Ischemia is closely associated with the four-hour cutoff. In prolonged episodes, corporal aspiration is far less effective, and emergent surgical intervention may be the only treatment available to prevent irreversible penile damage.30
Interestingly, hydroxyurea may have some efficacy in reducing the incidence of priapism in SCD patients. Sildenafil and tadalafil, acting as PDE-5 inhibitors, both significantly reduce the incidence of priapism, up to 92% in some studies. Unfortunately, when patients discontinued these PDE-5 inhibitors, they experienced episodes of priapism with significantly increased frequency and duration than baseline. Exercise may be a safer alternative; even climbing a staircase may resolve some episodes of priapism in the ED.30
While less common than in adults, pediatric patients with SCD also may experience priapism. These cases should be evaluated urgently by urology, given the typically poor reporting involved and the potentially devastating long-term consequences.30
Associated Conditions
SCD is associated with a wide range of chronic conditions and comorbidities. These patients may develop pulmonary hypertension, chronic renal disease, leg ulcers, sleep disturbance, anxiety, and retinopathy.1 All of these factors can contribute to a decreased quality of life and worsened patient outcomes.
Cardiac Comorbidities
As treatments for patients living with SCD have improved and life expectancies have increased, the mortality profile for this condition has evolved. In developed nations, pulmonary hypertension, sudden death syndrome, and diastolic dysfunction now are the leading causes of mortality for patients with SCD.31 Up to 33% of SCD patients may develop pulmonary hypertension. It has been suggested that this, in combination with chronic hemolytic anemia and microvascular vaso-occlusive crises, lead to myocardial scarring and left ventricular diastolic dysfunction.31 This combination of pulmonary hypertension and heart failure with preserved ejection fraction (HFpEF) has been shown in animal models. Ultimately, this may lead to further cardiac dysfunction, including a prolonged QT interval and even sudden death. Unfortunately, specific therapies for managing cardiac issues in SCD patients are “non-existent because the necessary clinical trials have not been conducted.”31 Emergency physicians should have a low threshold to suspect cardiac events in SCD patients. An electrocardiogram (ECG) should be obtained to assess the possibility of arrhythmias secondary to chronic heart disease in patients with palpitations or other suggestive symptoms, and echocardiograms may be used to evaluate diastolic function. When possible, patients with SCD should establish care with a cardiologist.
Renal Comorbidities
Renal function in patients with SCD declines with advancing age. Vaso-occlusive crises can induce ischemia in the kidneys, ultimately leading to the development of chronic kidney disease (CKD). Other processes associated with SCD, such as oxidative stress and hemolysis, also exacerbate this process. Approximately 30% of adult patients with SCD may experience a rapid decline in their estimated glomerular filtration rate (eGFR), defined as annual > 3 mL/min/1.73m2, a finding that is strongly associated with early mortality. Albuminuria can be identified with screening and is associated with eGFR decline. Interestingly, hydroxyurea appears to have a protective effect against the development of CKD in SCD. Chronic RBC transfusion also appears to delay the development of albuminuria.32 Emergency physicians should be mindful of the risk for renal insufficiency in this patient population and obtain laboratory studies, including a serum creatinine, prior to prescribing any medications that require renal dose adjustment. In suspected cases of worsening CKD, patients should be referred to nephrology.
Dermatological Comorbidities
Sickle cell leg ulcers (SCLUs) occur in 14% to 18% of patients with SCD in the United States. These painful, slow-healing ulcers can have significant negative impact on patients’ quality of life.33 They occur most often in male adults, and are associated with low hemoglobin and low fetal hemoglobin, often arising from a state of chronic hemolysis. Patients with sickle cell leg ulcers are at increased risk of pulmonary complications and have higher mortality. Data on SCLUs are very limited, but they appear to respond to standard protocols for wound care. However, these ulcers are unique in the extreme degrees of pain that they elicit in the patient, in their slow healing, and in the rate of recurrence.33
Psychiatric Comorbidities
In both adult and pediatric populations, SCD is strongly associated with higher rates of depression and anxiety than in the general population. More than 70% of adults with SCD display evidence of sleep disturbances as well.34 Patients with SCD should be routinely screened for these psychiatric comorbidities.
One of the dangers of treating this population is the long history of medical professionals stereotyping SCD patients as opioid-seeking.7 However, despite the prevalence of pain in this population, SCD patients are not more likely to develop addiction to pain medications than the general public.1 Because of repeated pain crises and exposure to analgesic medications, these patients may have higher than average tolerance for opioids, and individualized pain control plans with high doses of narcotics should not be taken as evidence of medication misuse or “drug seeking.” Emergency physicians should be wary of possible cognitive biases when treating these individuals, given the long history of mistrust that many of these patients have with the medical system.7,10
Ophthalmic Comorbidities
SCD can predispose patients to a wide variety of ophthalmic complications, including orbital wall infarction, orbital cellulitis, orbital compression syndrome, anterior segment ischemia leading to retinal detachment and vitreous hemorrhage, and a number of other pathologies. Ophthalmology should be consulted urgently for patients with SCD who present with any significant form of ocular involvement.35 SCD patients should have regular ophthalmologic examinations every one to two years starting at age 10 years.1
Chronic Treatment Regimens
Transfusion
The American Society of Hematology’s 2020 guidelines for SCD suggest that an extended red cell antigen profile be obtained for all patients with SCD at the earliest possible time, preferably before any transfusions are required. For reasons that remain unclear, SCD patients have the highest RBC alloimmunization incidence of any patient population. Therefore, it is imperative that prophylactic RBC antigen matching be done for these patients whenever possible.36 When possible, automated exchange transfusion should be performed, since simple transfusion can lead to iron overload and hyperviscosity.36 Even with this, patients with SCD who undergo chronic transfusion therapy likely would benefit from an MRI to establish their liver iron content every one to two years, in addition to serial monitoring of their ferritin levels.36
Hydroxyurea
For 20 years, hydroxyurea was the sole pharmacological tool for the long-term treatment of SCD approved by the U.S. Food and Drug Administration (FDA).1 Hydroxyurea works by inhibiting the ribonucleotide reductase (RNR) enzyme, a crucial step during the process of deoxyribonucleic acid (DNA) replication in a cell’s replication cycle. By reducing a free radical within the RNR’s structure, hydroxyurea prevents the formation of daughter strands of DNA from the original. The cell is forced into the S-phase checkpoint, halting mitosis and effectively forcing the entire cell cycle to pause. As the cell consumes more resources during this pause, the cytotoxic effects from hydroxyurea increase until, ultimately, cell death results. This unique property also allows hydroxyurea to be an effective antitumor treatment, in addition to treatment for SCD. Interestingly, this cytotoxic effect does not seem to negatively affect SCD patients; there is no evidence that hydroxyurea significantly damages the DNA of this patient population.37 In SCD patients specifically, hydroxyurea’s therapeutic effects occur by increasing fetal hemoglobin levels through a process that still is not completely understood.38 The degree of response may vary significantly between patients, inducing fetal hemoglobin levels from 2% to more than 30%. Fetal hemoglobin does not polymerize in the same manner as hemoglobin S and, therefore, does not cause the same sickle-shaped deformation of erythrocytes. Fetal hemoglobin is made preferentially in utero, but it usually is replaced with the production of regular hemoglobin following birth. Increasing the proportion of fetal hemoglobin relative to hemoglobin S in patients with SCD decreases the tendency of the RBCs to deform into a sickle shape.39
Hydroxyurea remains the first-line treatment for SCD and should be initiated at approximately 9 months of age.1 A 2020 study in the New England Journal of Medicine concluded that hydroxyurea with a gradual dose escalation is more effective than a fixed-dose hydroxyurea regimen. In the study of children in sub-Saharan Africa with SCD, researchers found that starting a hydroxyurea dose from 25 ± 5 mg/kg per day, and gradually increasing every two months to a maximum of 35 mg/kg per day, demonstrated significantly better clinical outcomes than a fixed-dose hydroxyurea regimen of 20 ± 5 mg/kg per day.40 The difference between the two groups was so stark, with the dose-escalation group experiencing 50% fewer SCD-related adverse events, that the researchers halted the trial early at 18 months.40 For chronic treatment of SCD, dose escalation of hydroxyurea is a safe and effective means of providing better clinical outcomes for patients.40 Any increased costs in laboratory monitoring are offset by the reduced number of adverse events these patients will experience.40 However, hydroxyurea remains severely underused — only 28% of eligible children are prescribed it, a statistic that is even worse in adults with SCD. This may be due to a combination of patients’ mistrust in the medical system and clinicians who are uncomfortable treating SCD.1 When appropriate, the emergency physician should suggest that patients discuss hydroxyurea with their primary physician.
Hydroxyurea’s side effects include abdominal pain, nausea, diarrhea, and the possibility of myelosuppression.1 Folic acid supplementation should be prescribed to patients taking hydroxyurea. Unfortunately, hydroxyurea’s inhibition of RNR potentially has teratogenic potential in pregnancy. This is particularly problematic in the United States, where African American women already are at significantly increased risk of miscarriage. In a self-reported study, researchers found that women who took hydroxyurea at conception and during pregnancy had a two-fold increased risk of miscarriage. Although the study was limited by recall bias, it suggests that hydroxyurea may be associated with an increased risk of miscarriage.41 When possible, hydroxyurea should be discontinued at least three months before conception.27 Even in male patients, hydroxyurea may have reproductive effects. There is conflicting evidence that long-term use of hydroxyurea may be associated with hypogonadism and reduced spermatogenesis.38
Other Treatments
In recent years, new treatments have emerged for the management of SCD. L-glutamine, an amino acid supplement, reduces RBC sickling by decreasing reactive oxygen species in erythrocytes. It may be prescribed as an alternative for patients unable to take hydroxyurea and was approved by the FDA for use in SCD patients age 5 years and older in July 2017.1,38 Similarly, crizanlizumab acts as an antibody against P-selectin, partially preventing adhesion to endothelial cells and thus reducing the vaso-occlusive pain crises that may be experienced with SCD.1 Crizanlizumab was approved by the FDA in November 2019 for the prophylactic reduction of pain crises in SCD patients 16 years of age and older.38
Hemopoietic stem cell transplantation (HSCT) has been a known treatment for SCD since 1984, and there are a number of studies examining the further possibilities of HSCT and/or allogenic bone marrow transplantation as treatment options.38 Unfortunately, the high financial barriers to these treatments, as well as the possibility of serious treatment-related complications such as graft vs. host disease (GVHD), currently limit the practical potential of these therapies for many patients.38
In late 2023, the FDA approved a novel treatment for SCD using clustered regularly interspaced short palindromic repeats (CRISPR) technology. The first approved product, exagamglogene autotemcel, is a one-time treatment that increases fetal hemoglobin by targeting the gene that inhibits fetal hemoglobin production in adults. While clinical trials showed increased fetal hemoglobin and clinical improvement in the symptoms and complications associated with SCD, the significant cost of the treatment (more than
$2 million at the time of this writing) and complex administration requirements, which include chemotherapy and hospitalization, may limit widespread adoption. Nonetheless, this gene-editing technology offers exciting possibilities for future SCD therapy.42,43
Access to Care
Because of the lifelong nature of SCD, it is important that these patients have long-term, comprehensive outpatient care by providers who are knowledgeable in the management of the disease. Pediatric patients especially require access to a wide range of preventive visits, long-term prophylactic treatments including hydroxyurea and exchange transfusions, and close follow-up.44 Only two-thirds of pediatric SCD patients have access to this care, which is further impaired in 80% of cases by ineffective coordination between their primary care provider and the multiple specialists that are required. Other barriers, including logistical challenges, difficulty obtaining insurance, difficulty in taking time off work, treatment delays, poor communication, and “maltreatment or marginalization from healthcare providers,” also can contribute to a reduced quality of life for these patients.44
Unfortunately, for many adult patients with SCD who present to the ED, this trend continues. One survey of SCD patients who presented to the ED found nearly half reported their annual household income as $15,000 or less.45 Other social determinants of health, such as pain crises limiting employment opportunities and the ability to acquire private healthcare, further limit many SCD patients from obtaining proper follow-up and care with a long-term provider.45 In the ED setting, these barriers can be difficult for physicians to overcome. One option includes referring affected individuals to organizations such as the Sickle Cell Disease Association of America, which can provide outreach and services to affected individuals. Expanding the use of electronic medical records to better develop communication and foster trust with these patients, in a respectful and culturally appropriate manner, also may facilitate improved outcomes for this patient population.44
While the vast majority of patients with SCD make one or fewer visits to an ED a year, there is a small group of patients who visit EDs more often. In some cases, this may be a sign of worsening disease. In others, the patient may not have a strong link to appropriate care and use the ED as a source of pain medication. Patients in this latter group should be referred to a compassionate, dedicated physician when possible, and connected with peer groups through agencies such as the Sickle Cell Disease Association of America.
Conclusion
SCD is a complex condition with diverse potential complications that may pose a significant challenge to unprepared clinicians. In the ED setting, physicians should be aware of the life-threatening pathologies that can affect patients with SCD.
REFERENCES
- Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: A review. JAMA 2022;328:57-68.
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet 2017;390:311-323.
- Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med 2017;376:1561-1573.
- Rees DC, Williams TN, Gladwin, MT. Sickle-cell disease. Lancet 2010;376:2018-2031.
- Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers 2018;4:1-22.
- DiMartino LD, Baumann AA, Hsu LL, et al. The sickle cell disease implementation consortium: Translating evidence-based guidelines into practice for sickle cell disease. Am J Hematol 2018;93:E391-E395.
- Ogu UO, Badamosi NU, Camacho PE, et al. Management of sickle cell disease complications beyond acute chest syndrome. J Blood Med 2021;12:101-114.
- Abdallah K, Buscetta A, Cooper K, et al. Emergency department utilization for patients living with sickle cell disease: Psychosocial predictors of health care behaviors. Ann Emerg Med 2020;76:S56-S63.
- Stevens DL, Hix M, Gildon BL. Crizanlizumab for the prevention of vaso-occlusive pain crises in sickle cell disease. J Pharm Technol 2021;37:209-215.
- Lovett PB, Sule HP, Lopez BL. Sickle cell disease in the emergency department. Hematol/Oncol Clin North Am 2017;31:1061-1079.
- Yusuf HR, Atrash HK, Grosse SD, et al. Emergency department visits made by patients with sickle cell disease: A descriptive study, 1999-2007. Am J Prev Med 2010;38(4 Suppl):S536-S541.
- Darbari DS, Sheehan VA, Ballas SK. The vaso‐occlusive pain crisis in sickle cell disease: Definition, pathophysiology, and management. Eur J Haematol 2020;105:237-246.
- Cline DM, Silva S, Freiermuth CE, et al. Emergency department (ED), ED observation, day hospital, and hospital admissions for adults with sickle cell disease. West J Emerg Med 2018;19:311.
- Lanzkron S, Little J, Wang, H, et al. Treatment of acute pain in adults with sickle cell disease in an infusion center versus the emergency department: A multicenter prospective cohort study. Ann Intern Med 2021;174:1207-1213.
- Alshurafa A, Soliman AT, De Sanctis V, et al. Clinical and epidemiological features and therapeutic options of avascular necrosis in patients with sickle cell disease (SCD): A cross-sectional study. Acta Biomedica 2023;94:e2023198.
- Ejindu VC, Hine AL, Mashayekhi M, et al. Musculoskeletal manifestations of sickle cell disease. Radiographics 2007;27:1005-1021.
- Ochocinski D, Dalal M, Black LV, et al. Life-threatening infectious complications in sickle cell disease: A concise narrative review. Front Pediatr 2020;8:38.
- Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: A review. Int J Infect Dis 2010;14:e2-e12.
- Eastep TG, Kendsersky RM, Zook J, Moore A. Penicillin prophylaxis in patients with sickle cell disease beyond age 5 years. The Journal of Pediatric Pharmacology and Therapeutics 2023;28:519-523.
- Cannas G, Merazga S, Virot E. Sickle cell disease and infections in high-and low-income countries. Mediterr J Hematol Infect Dis 2019;11:e2019042.
- Koehl JL, Koyfman A, Hayes BD, Long B. High risk and low prevalence diseases: Acute chest syndrome in sickle cell disease. Am J Emerg Med 2022;58:235-244.
- Novelli EM, Gladwin MT. Crises in sickle cell disease. Chest 2016;149:1082-1093.
- Paul RN, Castro OL, Aggarwal A, Oneal PA. Acute chest syndrome: Sickle cell disease. Eur J Haematol 2011;87:191-207.
- Verduzco LA, Nathan DG. Sickle cell disease and stroke. Blood 2009;114:5117-5125.
- Abdullahi SU, Jibir BW, Bello-Manga, H, et al. Hydroxyurea for primary stroke prevention in children with sickle cell anaemia in Nigeria (SPRING): A double-blind, multicentre, randomised, phase 3 trial. Lancet Haematol 2022;9:e26-e37.
- Strouse JJ, Lanzkron S, Urrutia V. The epidemiology, evaluation and treatment of stroke in adults with sickle cell disease. Expert Rev Hematol 2011;4:597-606.
- Jain D, Atmapoojya P, Colah R, Lodha P. Sickle cell disease and pregnancy. Mediterr J Hematol Infect Dis 2019;11:e2019040.
- Noubiap JJ, Temgoua MN, Tankeu R, et al. Sickle cell disease, sickle trait and the risk for venous thromboembolism: A systematic review and meta-analysis. Thrombosis J 2018;16:1-8.
- Chinegwundoh FI, Smith S, Anie KA. Treatments for priapism in boys and men with sickle cell disease. Cochrane Database Syst Rev 2020;4:CD004198.
- Idris IM, Burnett AL, DeBaun MR. Epidemiology and treatment of priapism in sickle cell disease. Hematology Am Soc Educ Program 2022;2022:450-458.
- Wood KC, Gladwin MT, Straub AC. Sickle cell disease: At the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2020;106:562-568.
- Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol 2022;18:361-377.
- Monfort JB, Senet P. Leg ulcers in sickle-cell disease: Treatment update. Adv Wound Care (New Rochelle) 2020;9:348-356.
- Wallen GR, Minniti CP, Krumlauf M, et al. Sleep disturbance, depression and pain in adults with sickle cell disease. BMC Psychiatry 2014;14:1-8.
- Elagouz M, Jyothi S, Gupta B, et al. Sickle cell disease and the eye: Old and new concepts. Surv Ophthalmol 2010;55:359-377.
- Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv 2020;4:327-355.
- Musiałek MW, Rybaczek D. Hydroxyurea—The good, the bad and the ugly. Genes (Basel) 2021;12:1096.
- Salinas Cisneros G, Thein SL. Recent advances in the treatment of sickle cell disease. Front Physiol 2020;11:435.
- Pule GD, Mowla S, Novitzky N, et al. A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev Hematol 2015;8:669-679.
- John CC, Opoka RO, Latham TS, et al. Hydroxyurea dose escalation for sickle cell anemia in Sub-Saharan Africa. N Engl J Med 2020;382:2524-2533.
- Kroner BL, Hankins JS, Pugh N, et al; Sickle Cell Disease Implementation Consortium. Pregnancy outcomes with hydroxyurea use in women with sickle cell disease. Am J Hematol 2022;97:603-612.
- Walker J. FDA approves world’s first Crispr gene-editing drug for sickle-cell disease. The Wall Street Journal. Published Dec. 8, 2023. https://www.wsj.com/tech/biotech/fda-approves-worlds-first-crispr-gene-editing-drug-for-sickle-cell-disease-8c65fbb3?st=108f5dpjox3eee6&reflink=desktopwebshare_permalink
- Sharma A, Boelens JJ, Cancio M, et al. CRISPR-Cas9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. N Engl J Med 2023;389:820-832.
- Jacob E, Childress C, Nathanson JD. Barriers to care and quality of primary care services in children with sickle cell disease. J Adv Nurs 2016;72:1417-1429.
- Linton EA, Goodin DA, Hankins JS, et al. A survey-based needs assessment of barriers to optimal sickle cell disease care in the emergency department. Ann Emerg Med 2020;76:S64-S72.
Sickle cell disease is a complex condition with diverse potential complications. In the emergency department setting, physicians should be aware of the life-threatening pathologies that can affect patients with sickle cell disease.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.